
Abstract 抽象
Organisms are defined by the information encoded in their genomes, and since the origin of life this information has been encoded using a two-base-pair genetic alphabet (A–T and G–C). In vitro, the alphabet has been expanded to include several unnatural base pairs (UBPs)1,2,3. We have developed a class of UBPs formed between nucleotides bearing hydrophobic nucleobases, exemplified by the pair formed between d5SICS and dNaM (d5SICS–dNaM), which is efficiently PCR-amplified1 and transcribed4,5 in vitro, and whose unique mechanism of replication has been characterized6,7. However, expansion of an organism’s genetic alphabet presents new and unprecedented challenges: the unnatural nucleoside triphosphates must be available inside the cell; endogenous polymerases must be able to use the unnatural triphosphates to faithfully replicate DNA containing the UBP within the complex cellular milieu; and finally, the UBP must be stable in the presence of pathways that maintain the integrity of DNA. Here we show that an exogenously expressed algal nucleotide triphosphate transporter efficiently imports the triphosphates of both d5SICS and dNaM (d5SICSTP and dNaMTP) into Escherichia coli, and that the endogenous replication machinery uses them to accurately replicate a plasmid containing d5SICS–dNaM. Neither the presence of the unnatural triphosphates nor the replication of the UBP introduces a notable growth burden. Lastly, we find that the UBP is not efficiently excised by DNA repair pathways. Thus, the resulting bacterium is the first organism to propagate stably an expanded genetic alphabet.
生物体是由其基因组中编码的信息定义的,自生命起源以来,这些信息一直使用两个碱基对的遗传字母表(A-T 和 G-C)进行编码。在体外,字母表已扩展到包括几个非天然碱基对 (UBP)1,2,3。我们开发了一类在带有疏水核碱基的核苷酸之间形成的 UBP,例如 d5SICS 和 dNaM 之间形成的一对 (d5SICS-dNaM),它在体外被有效地 PCR 扩增1 并转录4,5,并且其独特的复制机制已被表征6,7.然而,生物体遗传字母表的扩展带来了前所未有的新挑战:非天然的核苷三磷酸盐必须在细胞内可用;内源性聚合酶必须能够使用非天然三磷酸盐在复杂的细胞环境中忠实地复制含有 UBP 的 DNA;最后,UBP 必须在存在维持 DNA 完整性的通路的情况下保持稳定。在这里,我们表明外源表达的藻类核苷酸三磷酸转运蛋白有效地将 d5SICS 和 dNaM 的三磷酸盐(d5SICSTP 和 dNaMTP)输入到大肠杆菌中,并且内源复制机制使用它们来准确复制含有 d5SICS-dNaM 的质粒。非天然三磷酸盐的存在和 UBP 的复制都没有带来显著的生长负担。最后,我们发现 UBP 不能被 DNA 修复途径有效切除。因此,所得细菌是第一个稳定繁殖扩展的遗传字母表的生物体。
Similar content being viewed by others
其他人正在查看类似内容

Main 主要
To make the unnatural triphosphates available inside the cell, we previously suggested using passive diffusion of the free nucleosides into the cytoplasm followed by their conversion to the corresponding triphosphate via the nucleoside salvage pathway8. Although we have shown that analogues of d5SICS and dNaM are phosphorylated by the nucleoside kinase from Drosophila melanogaster8, monophosphate kinases are more specific9, and in E. coli we found that overexpression of the endogenous nucleoside diphosphate kinase results in poor growth. As an alternative, we focused on the nucleotide triphosphate transporters (NTTs) of obligate intracellular bacteria and algal plastids10,11,12,13,14. We expressed eight different NTTs in E. coli C41(DE3)15,16,17 and measured the uptake of [α-32P]-dATP as a surrogate for the unnatural triphosphates (Extended Data Fig. 1). We confirmed that [α-32P]-dATP is efficiently transported into cells by the NTTs from Phaeodactylum tricornutum (PtNTT2)18 and Thalassiosira pseudonana (TpNTT2)18. Although NTTs from Protochlamydia amoebophila (PamNTT2 and PamNTT5)15 also import [α-32P]-dATP, PtNTT2 showed the most activity, and both it and TpNTT2 are known to have broad specificity18, making them the most promising NTTs for further characterization.
为了使非天然的三磷酸盐在细胞内可用,我们之前建议将游离核苷被动扩散到细胞质中,然后通过核苷补救途径将它们转化为相应的三磷酸盐8。尽管我们已经证明 d5SICS 和 dNaM 的类似物被黑腹果蝇8 的核苷激酶磷酸化,但单磷酸激酶更具特异性9,在大肠杆菌中,我们发现内源性核苷二磷酸激酶的过表达导致生长不良。作为替代方案,我们专注于专性细胞内细菌和藻类质体的核苷酸三磷酸转运蛋白 (NTT)10,11,12,13,14。我们在大肠杆菌 C41 (DE3) 15、16、17 中表达了八种不同的 NTT,并测量了 [α-32 P]-dATP 作为非天然三磷酸盐替代物的摄取(扩展数据图 1)。我们证实 [α-32P]-dATP 被 NTT 从三角指螨 (PtNTT2)18 和假海蜥 (TpNTT2)18 的 NTT 有效地转运到细胞中。尽管来自嗜变形虫原体的 NTT(PamNTT2 和 PamNTT5)15 也输入 [α-32 P]-dATP,但 PtNTT2 显示出最强的活性,并且已知它和 TpNTT2 都具有广泛的特异性18,使其成为最有希望用于进一步表征的 NTT。
Transport via an NTT requires that the unnatural triphosphates are sufficiently stable in culture media; however, preliminary characterization of d5SICSTP and dNaMTP indicated that decomposition occurs in the presence of actively growing E. coli (Extended Data Fig. 2). Similar behaviour was observed with [α-32P]-dATP, and the dephosphorylation products detected by thin-layer chromatography (TLC) for [α-32P]-dATP, or by high-performance liquid chromatography (HPLC) and matrix-assisted laser desorption/ionization (MALDI) for d5SICSTP and dNaMTP, suggest that decomposition is mediated by phosphatases. As no degradation was observed upon incubation in spent media, decomposition seems to occur within the periplasm. No increase in stability was observed in cultures of single-gene-deletion mutants of E. coli BW25113 lacking a specific periplasmic phosphatase19 (as identified by the presence of a Sec-type amino-terminal leader sequence), including phoA, ushA, appA, aphA, yjjX, surE, yfbR, yjjG, yfaO, mutT, nagD, yggV, yrfG or ymfB, suggesting that decomposition results from the activity of multiple phosphatases. However, the extracellular stability of [α-32P]-dATP was significantly greater when 50 mM potassium phosphate (KPi) was added to the growth medium (Extended Data Fig. 3). Thus, we measured [α-32P]-dATP uptake from media containing 50 mM KPi after induction of the transporter with isopropyl-β-d-thiogalactoside (IPTG) (Extended Data Fig. 4). Although induction with 1 mM IPTG resulted in slower growth, consistent with the previously reported toxicity of NTTs17, it also resulted in maximal [α-32P]-dATP uptake. Thus, after addition of 1 mM IPTG, we analysed the extracellular and intracellular stability of [α-32P]-dATP as a function of time (Extended Data Fig. 5). Cells expressing PtNTT2 were found to have the highest levels of intracellular [α-32P]-dATP, and although both extra- and intracellular dephosphorylation was still observed, the ratio of triphosphate to dephosphorylation products inside the cell remained roughly constant, indicating that the extracellular concentrations and PtNTT2-mediated influx are sufficient to compensate for intracellular decomposition.
通过 NTT 运输要求非天然三磷酸盐在培养基中足够稳定;然而,d5SICSTP 和 dNaMTP 的初步表征表明,分解发生在活跃生长的大肠杆菌存在下(扩展数据图 2)。使用 [α-32P]-dATP 观察到类似的行为,通过薄层色谱 (TLC) 检测的 [α-32P]-dATP,或通过高效液相色谱 (HPLC) 和基质辅助激光解吸/电离 (MALDI) 检测的 d5SICSTP 和 dNaMTP 的去磷酸化产物,表明分解是由磷酸酶介导的。由于在用过的培养基中孵育时没有观察到降解,因此似乎在周质内发生分解。在缺乏特异性周质磷酸酶19(通过存在 Sec 型氨基末端前导序列鉴定)的大肠杆菌单基因缺失突变体BW25113培养物中未观察到稳定性增加,包括 phoA、ushA、appA、aphA、yjjX、surE、yfbR、yjjG、yfaO、mutT、nagD、yggV、yrfG 或ymfB,表明分解是由多种磷酸酶的活性引起的。然而,当向生长培养基中添加 50 mM 磷酸钾 (KPi) 时,[α-32P]-dATP 的细胞外稳定性显着更高(扩展数据图 3)。因此,我们测量了异丙基-β-d-硫代半乳糖苷 (IPTG) 诱导转运蛋白后含有 50 mM KPi 的培养基中 [α-32P]-dATP 的摄取(扩展数据图 4)。 尽管用 1 mM IPTG 诱导导致生长变慢,与先前报道的 NTTs毒性一致 17,但它也导致 [α-32 P]-dATP 摄取最大。因此,在添加 1 mM IPTG 后,我们分析了 [α-32P]-dATP 的细胞外和细胞内稳定性随时间的变化(扩展数据图 5)。发现表达 PtNTT2 的细胞具有最高水平的细胞内 [α-32P]-dATP,尽管仍观察到细胞外和细胞内去磷酸化,但细胞内三磷酸盐与去磷酸化产物的比率保持大致恒定,表明细胞外浓度和 PtNTT2 介导的内流足以补偿细胞内分解。
Likewise, we found that the addition of KPi increased the extracellular stability of d5SICSTP and dNaMTP (Extended Data Fig. 2), and when a stationary phase culture was diluted 100-fold into fresh media, the half-lives of both unnatural triphosphates (initial concentrations of 0.25 mM) were found to be approximately 9 h, which seemed sufficient for our purposes. Thirty minutes after their addition to the media, neither of the unnatural triphosphates was detected in cells expressing TpNTT2; in contrast, 90 μM of d5SICSTP and 30 μM of dNaMTP were found in the cytoplasm of cells expressing PtNTT2 (Fig. 1b). Although intracellular decomposition was still apparent, the intracellular concentrations of intact triphosphate are significantly above the sub-micromolar KM values of the unnatural triphosphates for DNA polymerases20, setting the stage for replication of the UBP in a living bacterial cell.
同样,我们发现添加 KPi 增加了 d5SICSTP 和 dNaMTP 的细胞外稳定性(扩展数据图 2),当固定相培养物稀释到新鲜培养基中 100 倍时,发现两种非天然三磷酸盐的半衰期(初始浓度为 0.25 mM)约为 9 小时,这似乎足以达到我们的目的。将它们添加到培养基中 30 分钟后,在表达 TpNTT2 的细胞中未检测到任何非天然三磷酸盐;相比之下,在表达 PtNTT2 的细胞质中发现了 90 μM 的 d5SICSTP 和 30 μM 的 dNaMTP(图 1b)。尽管细胞内分解仍然明显,但完整三磷酸盐的细胞内浓度显著高于 DNA 聚合酶20 的非天然三磷酸盐的亚微摩尔 KM 值,为 UBP 在活细菌细胞中的复制奠定了基础。
图 1:核苷三磷酸稳定性和输入。

a, Chemical structure of the d5SICS–dNaM UBP compared to the natural dG–dC base pair. b, Composition analysis of d5SICS and dNaM in the media (top) and cytoplasmic (bottom) fractions of cells expressing PtNTT2 after 30 min incubation; dA shown for comparison. 3P, 2P, 1P and 0P correspond to triphosphate, diphosphate, monophosphate and nucleoside, respectively; [3P] is the intracellular concentration of triphosphate. Error bars represent s.d. of the mean, n = 3.
a,d5SICS-dNaM UBP 与天然 dG-dC 碱基对的化学结构比较。b,孵育 30 分钟后表达 PtNTT2 的细胞的培养基(上图)和细胞质(下图)组分中 d5SICS 和 dNaM 的组成分析;dA 所示以供比较。3P、2P、1P 和 0P 分别对应于三磷酸盐、二磷酸盐、单磷酸盐和核苷;[3P] 是三磷酸盐的细胞内浓度。误差线表示平均值的 s.d.,n = 3。
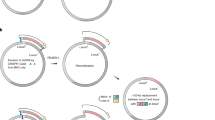
The replication of DNA containing d5SICS-dNaM has been validated in vitro with different polymerases, primarily family A polymerases, such as the Klenow fragment of E. coli DNA polymerase I (pol I)20,21. As the majority of the E. coli genome is replicated by pol III, we engineered a plasmid to focus replication of the UBP to pol I. Plasmid pINF (the information plasmid) was constructed from pUC19 using solid-phase DNA synthesis and circular-extension PCR to replace the dA–dT pair at position 505 with dNaM paired opposite an analogue of d5SICS (dTPT322) (Fig. 2a, b). This positions the UBP 362 bp downstream of the ColE1 origin of replication where leading-strand replication is mediated by pol I23, and within the TK-1 Okazaki processing site24, where lagging-strand synthesis is also expected to be mediated by pol I. Synthetic pINF was constructed using the d5SICS analogue because it should be efficiently replaced by d5SICS if replication occurs in vivo, making it possible to differentiate in vivo replicated pINF from synthetic pINF.
含有 d5SICS-dNaM 的 DNA 的复制已使用不同的聚合酶(主要是 A 家族聚合酶)在体外得到验证,例如大肠杆菌 DNA 聚合酶 I (pol I) 的 Klenow 片段20,21。由于大部分大肠杆菌基因组由 pol III 复制,我们设计了一个质粒以将 UBP 的复制集中在 pol I。质粒 pINF(信息质粒)是使用固相 DNA 合成和环状延伸 PCR 从 pUC19 构建的,以替换 dNaM 在 505 位的 dA-dT 对,与 d5SICS 的类似物 (dTPT322) 对对(图 2a, 这将 UBP 定位在 ColE1 复制起点下游 362 bp,其中前导链复制由 pol I23 介导,在 TK-1 冈崎加工位点24 内,滞后链合成也有望由 pol I 介导。合成 pINF 是使用 d5SICS 类似物构建的,因为如果在体内发生复制,它应该被 d5SICS 有效地取代,从而可以区分体内复制的 pINF 和合成的 pINF。
图 2:细胞内 UBP 复制。

a, Structure of pACS and pINF. dX and dY correspond to dNaM and a d5SICS analogue22 that facilitated plasmid construction (see Methods). cloDF, origin of replication; Sm, streptomycin resistance gene; AmpR, ampicillin resistance gene; ori, ColE1 origin of replication; lacZα, β-galactosidase fragment gene. b, Overview of pINF construction. A DNA fragment containing the unnatural nucleotide was synthesized via solid-phase DNA synthesis and then used to assemble synthetic pINF via circular-extension PCR29. X, dNaM; Y′, dTPT3 (an analogue of d5SICS22); y, d5SICS (see text). Colour indicates regions of homology. The doubly nicked product was used directly to transform E. coli harbouring pACS. c, The addition of d5SICSTP and dNaMTP eliminates a growth lag of cells harbouring pINF. EP, electroporation. Error bars represent s.d. of the mean, n = 3. d, LC-MS/MS total ion chromatogram of global nucleoside content in pINF and pUC19 recorded in dynamic multiple reaction monitoring (DMRM) mode. pINF and pUC19 (control) were propagated in E. coli in the presence or absence of unnatural triphosphates, and with or without PtNTT2 induction. The inset shows a 100-fold expansion of the mass-count axis in the d5SICS region. e, Biotinylation only occurs in the presence of the UBP, the unnatural triphosphates and transporter induction. After growth, pINF was recovered, and a 194-nucleotide region containing the site of UBP incorporation (nucleotides 437–630) was amplified and biotinylated. B, biotin; SA, streptavidin. The natural pUC19 control plasmid was prepared identically to pINF. A 50-bp DNA ladder is shown to the left. f, Sequencing analysis demonstrates retention of the UBP. An abrupt termination in the Sanger sequencing reaction indicates the presence of UBP incorporation (site indicated with arrow).
a, pACS 和 pINF 的结构。dX 和 dY 对应于 dNaM 和促进质粒构建的 d5SICS 类似物22(参见方法)。cloDF,复制起点;Sm, 链霉素耐药基因;AmpR, 氨苄青霉素抗性基因;ori,ColE1 复制起点;lacZα,β-半乳糖苷酶片段基因。b,pINF 构建概述。通过固相 DNA 合成合成含有非天然核苷酸的 DNA 片段,然后用于通过环状延伸 PCR 组装合成的 pINF29。X,dNaM;Y′,dTPT3(d5SICS22 的类似物);y, d5SICS (见正文).颜色表示同源区域。双切口产物直接用于转化含有 pACS 的大肠杆菌。c,添加 d5SICSTP 和 dNaMTP 消除了携带 pINF 的细胞的生长滞后。EP,电穿孔。误差线表示平均值的 s.d.,n = 3。d,动态多反应监测 (DMRM) 模式下记录的 pINF 和 pUC19 中全局核苷含量的 LC-MS/MS 总离子流色谱图。pINF 和 pUC19 (对照) 在存在或不存在非天然三磷酸盐的情况下在大肠杆菌中繁殖,并且有或没有 PtNTT2 诱导。插图显示了 d5SICS 区域中质量计数轴的 100 倍扩展。e,生物素化仅在存在 UBP、非自然三磷酸盐和转运蛋白诱导的情况下发生。生长后,pINF 被回收,并且含有 UBP 掺入位点(核苷酸 437-630)的 194 个核苷酸区域被扩增和生物素化。B, 生物素;SA,链霉亲和素。 天然 pUC19 对照质粒的制备方式与 pINF 相同。左侧显示了一个 50 bp 的 DNA 分子量标准。f,测序分析显示 UBP 保留。Sanger 测序反应的突然终止表明存在 UBP 掺入(用箭头表示的位点)。
To determine whether E. coli can use the imported unnatural triphosphates to stably propagate pINF, C41(DE3) cells were first transformed with a pCDF-1b plasmid encoding PtNTT2 (hereafter referred to as pACS, for accessory plasmid, Fig. 2a) and grown in media containing 0.25 mM of both unnatural triphosphates, 50 mM KPi and 1 mM IPTG to induce transporter production. Cells were then transformed with pINF, and after a 1-h recovery period, cultures were diluted tenfold with the same media supplemented with ampicillin, and growth was monitored via culture turbidity (Extended Data Table 1). As controls, cells were also transformed with pUC19, or grown without either IPTG or without the unnatural triphosphates. Again, growth was significantly slower in the presence of IPTG, but the addition of d5SICSTP and dNaMTP resulted in only a slight further decrease in growth in the absence of pINF, and interestingly, it eliminated a growth lag in the presence of pINF (Fig. 2c), suggesting that the unnatural triphosphates are not toxic and are required for the efficient replication of pINF.
为了确定大肠杆菌是否可以使用输入的非天然三磷酸盐稳定繁殖 pINF,首先用编码 PtNTT2 的 pCDF-1b 质粒(以下简称 pACS,用于辅助质粒,图 2a)转化 C41(DE3) 细胞,并在含有 0.25 mM 非天然三磷酸盐、50 mM KPi 和 1 mM IPTG 的培养基中生长以诱导转运蛋白产生。然后用 pINF 转化细胞,在 1 小时恢复期后,用补充有氨苄青霉素的相同培养基将培养物稀释 10 倍,并通过培养物浊度监测生长(扩展数据表 1)。作为对照,细胞也用 pUC19 转化,或者在没有 IPTG 或没有非天然三磷酸盐的情况下生长。同样,在 IPTG 存在下生长明显缓慢,但在不存在 pINF 的情况下,添加 d5SICSTP 和 dNaMTP 仅导致生长略有进一步降低,有趣的是,它在 pINF 存在下消除了生长滞后(图 2c),表明非天然三磷酸盐无毒,是 pINF 有效复制所必需的。
To demonstrate the replication of pINF, we recovered the plasmid from cells after 15 h of growth. The introduction of the UBP resulted in a small (approximately twofold) reduction in the copy number of pINF, as gauged by its ratio to pACS (Extended Data Table 1); we determined that the plasmid was amplified 2 × 107-fold during growth (approximately 24 doublings) based on the amount of recovered plasmid and the transformation efficiency. To determine the level of UBP retention, the recovered plasmid was digested, dephosphorylated to single nucleosides, and analysed by liquid chromatography-tandem mass spectrometry (LC-MS/MS)25. Although the detection and quantification of dNaM were precluded by its poor fragmentation efficiency and low product ion counts over background, signal for d5SICS was clearly observable (Fig. 2d). External calibration curves were constructed using the unnatural nucleoside and validated by determining its ratio to dA in synthetic oligonucleotides (Extended Data Table 2). Using the resulting calibration curve, we determined the ratio of dA to d5SICS in recovered pINF was 1,106 to 1, which when compared to the expected ratio of 1,325 to 1, suggests the presence of approximately one UBP per plasmid. No d5SICS was detected in control experiments in which the transporter was not induced, or when the unnatural triphosphates were not added to the media, or when pUC19 was used instead of pINF (Fig. 2d, inset), demonstrating that its presence results from the replication of the UBP and not from misinsertion of the unnatural triphosphates opposite a natural nucleotide. Importantly, as the synthetic pINF contained an analogue of d5SICS, and d5SICS was only provided as a triphosphate added to the media, its presence in pINF confirms in vivo replication.
为了证明 pINF 的复制,我们在生长 15 小时后从细胞中回收质粒。UBP 的引入导致 pINF 的拷贝数小幅减少(大约两倍),通过其与 pACS 的比率来衡量(扩展数据表 1);我们确定质粒在生长过程中扩增 2 × 107 倍(大约 24 倍),基于回收质粒的量和转化效率。为了确定 UBP 保留水平,将回收的质粒消化,去磷酸化为单核苷,并通过液相色谱-串联质谱 (LC-MS/MS) 分析25。尽管 dNaM 的碎裂效率差且背景上的产物离子计数低,因此无法检测和定量,但可以清楚地观察到 d5SICS 的信号(图 2d)。使用非天然核苷构建外部校准曲线,并通过测定其与合成寡核苷酸中 dA 的比率进行验证(扩展数据表 2)。使用得到的校准曲线,我们确定回收的 pINF 中 dA 与 d5SICS 的比率为 1,106 比 1,与预期的 1,325 比 1 的比率相比,表明每个质粒存在大约一个 UBP。在未诱导转运蛋白的对照实验中,或者当非天然三磷酸盐未添加到培养基中时,或者当使用 pUC19 代替 pINF 时,未检测到 d5SICS(图 2d,插图),表明其存在是由于 UBP 的复制,而不是由于非天然三磷酸盐与天然核苷酸相对的错误插入。 重要的是,由于合成的 pINF 含有 d5SICS 的类似物,而 d5SICS 仅以添加到培养基中的三磷酸盐的形式提供,因此它在 pINF 中的存在证实了体内复制。
To independently confirm and quantify the retention of the UBP in the recovered plasmid, the relevant region was amplified by PCR in the presence of d5SICSTP and a biotinylated dNaMTP analogue4 (Fig. 2e). Analysis by streptavidin gel shift showed that 67% of the amplified DNA contained biotin. No shift was observed in control experiments where the transporter was not induced, or when unnatural triphosphates were not added, or when pUC19 was used instead of pINF, demonstrating that the shift results from the presence of the UBP. Based on a calibration curve constructed from the shifts observed with the amplification products of controlled mixtures of DNA containing dNaM or its fully natural counterpart (Methods and Extended Data Fig. 6), the observed gel shift corresponds to a UBP retention of 86%. Similarly, when the amplification product obtained with d5SICSTP and dNaMTP was analysed by Sanger sequencing in the absence of the unnatural triphoshates1,26,27, the sequencing chromatogram showed complete termination at the position of UBP incorporation, which with an estimated lower limit of read-through detection of 5%, suggests a level of UBP retention in excess of 95% (Fig. 2f). In contrast, amplification products obtained from pINF recovered from cultures grown without PtNTT2 induction, without added unnatural triphosphates, or obtained from pUC19 propagated under identical conditions, showed no termination. Overall, the data unambiguously demonstrate that DNA containing the UBP was replicated in vivo and allow us to estimate that replication occurred with fidelity (retention per doubling) of at least 99.4% (24 doublings; 86% retention; 0.99424 = 0.86). This fidelity corresponds to an error rate of approximately 10−3, which is comparable to the intrinsic error rate of some polymerases with natural DNA28.
为了独立确认和定量 UBP 在回收质粒中的保留,在 d5SICSTP 和生物素化 dNaMTP 类似物4 存在下通过 PCR 扩增相关区域(图 2e)。通过链霉亲和素凝胶转移分析表明,67% 的扩增 DNA 含有生物素。在未诱导转运蛋白、未添加非天然三磷酸盐或使用 pUC19 代替 pINF 的对照实验中未观察到变化,表明变化是由于 UBP 的存在造成的。根据根据使用含有 dNaM 或其完全天然对应物的 DNA 的受控混合物的扩增产物观察到的偏移构建的校准曲线(方法和扩展数据图 6),观察到的凝胶偏移对应于 86% 的 UBP 保留率。同样,当在没有非天然三分体 1,26,27 的情况下通过 Sanger 测序分析使用 d5SICSTP 和 dNaMTP 获得的扩增产物时,测序色谱图显示 UBP 掺入位置完全终止,估计通读检测下限为 5%,表明 UBP 保留水平超过 95%(图 2f).相比之下,从未进行 PtNTT2 诱导、未添加非天然三磷酸盐的培养物中回收的 pINF 中获得的扩增产物,或从相同条件下繁殖的 pUC19 中获得的扩增产物未显示终止。总体而言,数据明确表明含有 UBP 的 DNA 在体内被复制,并允许我们估计复制的保真度(每次加倍保留率)至少为 99。4%(24 次倍增;86% 保留率;0.99424 = 0.86)。这种保真度对应于大约 10-3 的错误率,这与某些具有天然 DNA 的聚合酶的内在错误率相当28。
The high retention of the UBP over a 15-h period of growth (approximately 24 doublings) strongly suggests that it is not efficiently excised by DNA repair pathways. To test further this hypothesis and to examine retention during prolonged stationary phase growth, we repeated the experiments, but monitored UBP retention, cell growth and unnatural triphosphate decomposition for up to 6 days without providing any additional unnatural triphosphates (Fig. 3 and Extended Data Fig. 7). At 15 and 19 h of growth, the cultures reached an optical density at 600 nm (OD600) of approximately 0.9 and 1.2, respectively, and both d5SICSTP and dNaMTP decomposed to 17–20% and 10–16% of their initial 0.25-mM concentrations (Extended Data Fig. 7a). In agreement with the experiments described above, retention of the UBP after 15 h was 97 ± 5% and >95%, as determined by gel shift and sequencing, respectively, and after 19 h it was 91 ± 3% and >95%. As the cultures entered stationary phase and the triphosphates decomposed completely, plasmid loss began to compete with replication (Extended Data Fig. 7b, c, d), but even then, retention of the UBP remained at approximately 45% and 15%, at days 3 and 6 respectively. Moreover, when d5SICS-dNaM was lost, it was replaced by dA–dT, which is consistent with the mutational spectrum of DNA pol I20. Finally, the shape of the retention versus time curve mirrors that of the growth versus time curve. Taken together, these data suggest that in the absence of unnatural triphosphates, the UBP is eventually lost by replication-mediated mispairing, and not from the activity of DNA repair pathways.
UBP 在 15 小时的生长期间(约 24 次倍增)的高保留率强烈表明它不能被 DNA 修复途径有效切除。为了进一步检验这一假设并检查长时间固定相生长期间的保留性,我们重复了实验,但监测了 UBP 保留、细胞生长和非自然三磷酸分解长达 6 天,而没有提供任何额外的非天然三磷酸盐(图 3 和扩展数据图 7)。在生长 15 小时和 19 小时时,培养物在 600 nm 处的光密度 (OD600) 分别达到约 0.9 和 1.2,d5SICSTP 和 dNaMTP 均分解至其初始 0.25 mM 浓度的 17-20% 和 10-16%(扩展数据图 7a)。与上述实验一致,通过凝胶转移和测序确定 15 小时后 UBP 的保留率分别为 97 ± 5% 和 >95%,19 小时后为 91 ± 3% 和 >95%。随着培养物进入固定期并且三磷酸盐完全分解,质粒损失开始与复制竞争(扩展数据图 7b、c、d),但即便如此,UBP 的保留率分别保持在大约 45% 和 15%,分别在第 3 天和第 6 天。此外,当 d5SICS-dNaM 丢失时,它被 dA-dT 取代,这与 DNA pol I20 的突变谱一致。最后,保留率与时间曲线的形状反映了增长与时间曲线的形状。综上所述,这些数据表明,在没有非天然三磷酸盐的情况下,UBP 最终会因复制介导的错配对而丢失,而不是因 DNA 修复途径的活性而丢失。
图 3:UBP 的细胞内稳定性。

E. coli C41(DE3) pACS was transformed with pINF and grown after a single dose of d5SICSTP and dNaMTP was provided in the media. UBP retention in recovered pINF, OD600, and relative amount of d5SICSTP and dNaMTP in the media (100% = 0.25 mM), were determined as a function of time. Error bars represent s.d. of the mean, n = 3.
大肠杆菌用 pINF 转化 C41 (DE3) pACS,并在单剂量 d5SICSTP 后生长,并在培养基中提供 dNaMTP。将回收的 pINF 中的 UBP 保留率、OD600 以及培养基中 d5SICSTP 和 dNaMTP 的相对量 (100% = 0.25 mM) 确定为时间的函数。误差线表示平均值的 s.d.,n = 3。
We have demonstrated that PtNTT2 efficiently imports d5SICSTP and dNaMTP into E. coli and that an endogenous polymerase, possibly pol I, efficiently uses the unnatural triphosphates to replicate DNA containing the UBP within the cellular environment with reasonable efficiency and fidelity. Moreover, the UBP appears stable during both exponential and stationary phase growth despite the presence of all DNA repair mechanisms. Remarkably, although expression of PtNTT2 results in a somewhat reduced growth rate, neither the unnatural triphosphates nor replication of the UBP results in significant further reduction in growth. The resulting bacterium is the first organism that stably harbours DNA containing three base pairs. In the future, this organism, or a variant with the UBP incorporated at other episomal or chromosomal loci, should provide a synthetic biology platform to orthogonally re-engineer cells, with applications ranging from site-specific labelling of nucleic acids in living cells to the construction of orthogonal transcription networks and eventually the production and evolution of proteins with multiple, different unnatural amino acids.
我们已经证明,PtNTT2 有效地将 d5SICSTP 和 dNaMTP 输入到大肠杆菌中,并且内源性聚合酶(可能是 pol I)有效地利用非天然三磷酸在细胞环境中以合理的效率和保真度复制含有 UBP 的 DNA。此外,尽管存在所有 DNA 修复机制,但 UBP 在指数期和固定期生长期间都表现出稳定。值得注意的是,尽管 PtNTT2 的表达导致生长速率略有降低,但非自然的三磷酸盐和 UBP 的复制均不会导致生长显著进一步降低。所得细菌是第一个稳定地含有三个碱基对的 DNA 的生物体。将来,这种生物体或在其他游离型或染色体位点掺入 UBP 的变体应该提供一个合成生物学平台来对细胞进行正交重新设计,其应用范围从活细胞中核酸的位点特异性标记到构建正交转录网络,并最终产生和进化具有多个、 不同的非天然氨基酸。
Methods Summary 方法总结
To prepare electrocompetent C41(DE3) pACS cells, freshly transformed E. coli C41(DE3) pACS was grown overnight in 2 × YT medium (1.6% tryptone, 1% yeast extract, 0.5% NaCl) supplemented with streptomycin and KPi. After 100-fold dilution into the same medium and outgrowth at 37 °C to OD600 = 0.20, IPTG was added to induce expression of PtNTT2. After 40 min, cultures were rapidly cooled, washed with sterile water and resuspended in 10% glycerol. An aliquot of electrocompetent cells was mixed with pINF and electroporated. Pre-warmed 2 × YT medium containing streptomycin, IPTG and KPi was added, and an aliquot was diluted 3.3-fold in the same media supplemented with 0.25 mM each of dNaMTP and d5SICSTP. The resulting mixture was allowed to recover at 37 °C with shaking. After recovery, cultures were centrifuged. Spent media was analysed for nucleotide composition by HPLC (Extended Data Fig. 7a); cells were resuspended in fresh medium containing streptomycin, ampicillin, IPTG, KPi and 0.25 mM each of dNaMTP and d5SICSTP, and grown with shaking. At defined time points, OD600 was determined and aliquots were removed and centrifuged. Spent media were analysed for nucleotide composition, and pINF was recovered by spin column purification. UBP retention was characterized by LC-MS/MS, PCR amplification and gel electrophoresis, or sequencing, as described in the Methods.
为了制备电感受态 C41 (DE3) pACS 细胞,将新鲜转化的大肠杆菌 C41 (DE3) pACS 在补充有链霉素和 KPi 的 2 × YT 培养基(1.6% 胰蛋白胨、1% 酵母提取物、0.5% NaCl)中生长过夜。在相同的培养基中稀释 100 倍并在 37 °C 下生长至 OD600 = 0.20 后,加入 IPTG 以诱导 PtNTT2 的表达。40 分钟后,快速冷却培养物,用无菌水洗涤并重悬于 10% 甘油中。将等分试样的电感受态细胞与 pINF 混合并进行电穿孔。加入含有链霉素、IPTG 和 KPi 的预热 2 × YT 培养基,并在补充有 0.25 mM dNaMTP 和 d5SICSTP 的相同培养基中稀释 3.3 倍等分试样。将所得混合物在 37 °C 下振荡恢复。回收后,离心培养物。通过 HPLC 分析用过的培养基的核苷酸组成(扩展数据图 7a);将细胞重悬于含有链霉素、氨苄青霉素、IPTG、KPi 和 dNaMTP 和 d5SICSTP 各 0.25 mM 的新鲜培养基中,并摇动生长。在确定的时间点,测定 OD600,取出等分试样并离心。分析用过的培养基的核苷酸组成,并通过离心柱纯化回收 pINF。如 METHODS,LC-MS/MS、PCR 扩增和凝胶电泳或测序表征 UBP 保留。
Online Methods 在线方法
Materials 材料
2 × YT, 2 × YT agar, IPTG, ampicillin and streptomycin were obtained from Fisher Scientific. Ampicillin and streptomycin were used at 100 μg ml−1 and 50 μg ml−1, respectively. All pET-16b constructs containing the nucleotide transporters were kindly provided by I. Haferkamp (Technische Universität Kaiserslautern, Germany) with the exception of pET16b-RpNTT2, which along with the C41(DE3) E. coli strain, was provided by J. P. Audia (University of South Alabama, USA). Plasmids pUC19 and pCDF-1b were obtained from Thermo Scientific and EMD Millipore, respectively. Plasmids were purified using the PureLink Quick Plasmid DNA Miniprep Kit (Life Technologies). OneTaq, DeepVent, Q5 Hot Start High-Fidelity DNA Polymerases, and all restriction endonucleases were obtained from New England Biolabs. In general, PCR reactions were divided into multiple aliquots with one followed in real time using 0.5 × Sybr Green I (Life Technologies); following PCR, the aliquots were recombined, purified by spin column (DNA Clean and Concentrator-5; Zymo Research, Irvine, California, USA) with elution in 20 μl of water, then separated by agarose gel electrophoresis, followed by band excision and recovery (Zymoclean Gel DNA Recovery Kit), eluting with 20 μl of water unless stated otherwise. Polyacrylamide gels were stained with 1 × Sybr Gold (Life Technologies) for 30 min, agarose gels were cast with 1 × Sybr Gold. All gels were visualized using a Molecular Imager Gel Doc XR+ equipped with 520DF30 filter (Bio-Rad) and quantified with Quantity One software (Bio-Rad). The sequences of all DNA oligonucleotides used in this study are provided in Supplementary Information. Natural oligonucleotides were purchased from IDT (San Diego, California, USA). The concentration of dsDNA was measured by fluorescent dye binding (Quant-iT dsDNA HS Assay kit, Life Technologies) unless stated otherwise. The concentration of ssDNA was determined by UV absorption at 260 nm using a NanoDrop 1000 (Thermo Scientific). [α-32P]-dATP (25 µCi) was purchased from PerkinElmer (Shelton, Connecticut, USA). Polyethyleneimine cellulose pre-coated Bakerflex TLC plates (0.5 mm) were purchased from VWR. dNaM phosphoramidite, dNaM and d5SICS nucleosides were obtained from Berry & Associates Inc. (Dexter, Michigan, USA). Free nucleosides of dNaM and d5SICS (Berry & Associates) were converted to the corresponding triphosphates under Ludwig conditions30. After purification by anion exchange chromatography (DEAE Sephadex A-25) followed by reverse phase (C18) HPLC and elution through a Dowex 50WX2-sodium column, both triphosphates were lyophilized and kept at −20 °C until use. The d5SICSTP analogue dTPT3TP22 and the biotinylated dNaMTP analogue dmmo2SSBIOTP4 were made as reported previously. MALDI-TOF mass spectrometry (Applied Biosystems Voyager DE-PRO System 6008) was performed at the TSRI Center for Protein and Nucleic Acid Research.
2 × YT、2 × YT 琼脂、IPTG、氨苄青霉素和链霉素购自Fisher Scientific。氨苄青霉素和链霉素的用量分别为 100 μg ml-1 和 50 μg ml-1。所有含有核苷酸转运蛋白的 pET-16b 构建体均由 I. Haferkamp(德国凯泽斯劳滕工业大学)友情提供,但 pET16b-Rp NTT2 除外,它与 C41(DE3) 大肠杆菌菌株一起由 JP Audia(美国南阿拉巴马大学)提供。质粒 pUC19 和 pCDF-1b 分别从 Thermo Scientific 和 EMD Millipore 获得。使用 PureLink 快速质粒 DNA 小量制备试剂盒 (Life Technologies) 纯化质粒。OneTaq、DeepVent、Q5 热启动高保真 DNA 聚合酶和所有限制性核酸内切酶均购自 New England Biolabs。通常,将 PCR 反应分成多个等分试样,使用 0.5 × Sybr Green I (Life Technologies) 实时跟进一个;PCR 后,将等分试样重组,通过离心柱(DNA Clean 和 Concentrator-5;Zymo Research,Irvine,California,USA),用 20 μl 水洗脱,然后通过琼脂糖凝胶电泳分离,然后进行条带切除和回收(Zymoclean 凝胶 DNA 回收试剂盒),除非另有说明,否则用 20 μl 水洗脱。聚丙烯酰胺凝胶用 1 × Sybr Gold (Life Technologies) 染色 30 分钟,琼脂糖凝胶用 1 × Sybr Gold 灌注。使用配备 520DF30 滤光片 (Bio-Rad) 的 Molecular Imager Gel Doc XR+ 对所有凝胶进行可视化,并使用 Quantity One 软件 (Bio-Rad) 进行定量。本研究中使用的所有 DNA 寡核苷酸的序列在补充信息中提供。 天然寡核苷酸购自IDT(美国加利福尼亚州圣地亚哥)。除非另有说明,否则通过荧光染料结合(Quant-iT dsDNA HS 检测试剂盒,Life Technologies)测量 dsDNA 的浓度。使用 NanoDrop 1000 (Thermo Scientific) 在 260 nm 处通过紫外吸收测定 ssDNA 的浓度。[α-32P]-dATP (25 μCi) 购自 PerkinElmer (美国康涅狄格州谢尔顿)。聚乙烯亚胺纤维素预涂层 Bakerflex TLC 板 (0.5 mm) 购自 VWR。dNaM亚磷酰胺,dNaM和d5SICS核苷是从Berry & Associates Inc.(美国密歇根州的Dexter获得的。dNaM和d5SICS (Berry & Associates)的游离核苷在路德维希条件下转化为相应的三磷酸盐30。通过阴离子交换色谱 (DEAE Sephadex A-25) 纯化,然后进行反相 (C18) HPLC 纯化并通过 Dowex 50WX2 钠柱洗脱后,将两种三磷酸盐冻干并保持在 -20 °C 直至使用。d5SICSTP 类似物 dTPT3TP22 和生物素化 dNaMTP 类似物 dmmo2SSBIOTP4 如前所述制备。MALDI-TOF 质谱法(Applied Biosystems Voyager DE-PRO 系统 6008)在 TSRI 蛋白质和核酸研究中心进行。
Construction of NTT expression plasmids
NTT 表达质粒的构建
The PtNTT2 gene was amplified from plasmid pET-16b-PtNTT2 using primers PtNTT2-forward and PtNTT2-reverse; the TpNTT2 gene was amplified from plasmid pET-16b-TpNTT2 using primers TpNTT2-forward and TpNTT2-reverse. A linear fragment of pCDF-1b was generated using primers pCDF-1b-forward and pCDF-1b-reverse. All fragments were purified as described in Materials. The pCDF-1b fragment (100 ng, 4.4 × 10−14 mol) and either the PtNTT2 (78 ng, 4.4 × 10−14 mol) or TpNTT2 (85 ng, 4.4 × 10−14 mol) fragment were then assembled together using restriction-free circular polymerase extension cloning29 in 1 × OneTaq reaction buffer, MgSO4 adjusted to 3.0 mM, 0.2 mM of dNTP, and 0.02 U μl−1 of OneTaq DNA under the following thermal cycling conditions: initial denaturation (96 °C, 1 min); 10 cycles of denaturation (96 °C, 30 s), touchdown annealing (54 °C to 49.5 °C for 30 s (−0.5 °C per cycle)), extension of 68 °C for 5 min, and final extension (68 °C, 5 min). Upon completion, the samples were purified and used for heat-shock transformation of E. coli XL10. Individual colonies were selected on lysogeny broth (LB)-agar containing streptomycin, and assayed by colony PCR with primers PtNTT2-forward/reverse or TpNTT2-forward/reverse. The presence of the NTT genes was confirmed by sequencing and double digestion with ApaI/EcoO109I restriction endonucleases with the following expected pattern: pCDF-1b-PtNTT2 (2,546/2,605 bp), pCDF-1b-TpNTT2 (2,717/2,605 bp), pCDF-1b (1,016/2,605 bp). The complete nucleotide sequence of the pCDF-1b-PtNTT2 plasmid (pACS) is provided in Supplementary Information.
使用引物 PtNTT2-正向和 PtNTT2-反向从质粒 pET-16b-Pt NTT2 中扩增 PtNTT2 基因;使用引物 TpNTT2-forward 和 TpNTT2-reverse 从质粒 pET-16b-Tp NTT2 中扩增 TpNTT2 基因。使用引物 pCDF-1b-forward 和 pCDF-1b-reverse 生成 pCDF-1b 的线性片段。所有片段均按照材料中所述进行纯化。然后,使用无限制的环状聚合酶延伸克隆29 in 1 × OneTaq 反应缓冲液,将 pCDF-1b 片段(100 ng,4.4 × 10-14 mol)和 PtNTT2(78 ng,4.4 × 10-14 mol×)片段组装在一起,将 MgSO4 调节至 3.0 mM、0.2 mM dNTP。 和0.02 U μl-1 OneTaq DNA,在以下热循环条件下:初始变性(96 °C,1 分钟);10 个变性循环(96 °C,30 秒),着陆退火(54 °C 至 49.5 °C 持续 30 秒(每个循环 -0.5 °C)),68 °C 延伸 5 分钟,最后延伸(68 °C,5 分钟)。完成后,对样品进行纯化并用于大肠杆菌 XL10 的热休克转化。在含有链霉素的溶原肉汤 (LB)-琼脂上选择单个菌落,并使用引物 PtNTT2 正向/反向或 TpNTT2 正向/反向通过菌落 PCR 进行测定。通过用 ApaI/EcoO109I 限制性核酸内切酶进行测序和双重消化来确认 NTT 基因的存在,其预期模式如下:pCDF-1b-Pt NTT2 (2,546/2,605 bp)、pCDF-1b-Tp NTT2 (2,717/2,605 bp)、pCDF-1b (1,016/2,605 bp)。 pCDF-1b-Pt NTT2 质粒 (pACS) 的完整核苷酸序列在补充信息中提供。
Growth conditions to quantify nucleoside triphosphate uptake
量化核苷三磷酸摄取的生长条件
E. coli C41(DE3)16 freshly transformed with pCDF-1b-PtNTT2 was grown in 2 × YT with streptomycin overnight, then diluted (1:100) into fresh 2 × YT medium (1 ml of culture per uptake with [α-32P]-dATP; 2 ml of culture per uptake with d5SICSTP or dNaMTP) supplemented with 50 mM potassium phosphate (KPi) and streptomycin. A negative control with the inactive transporter pET-16b-RpNTT2, was treated identically except ampicillin was used instead of streptomycin. Cells were grown to an OD600 of approximately 0.6 and the NTT expression was induced by the addition of IPTG (1 mM). The culture was allowed to grow for another hour (final OD600 approximately 1.2) and then assayed directly for uptake as described below using a method adapted from a previous paper15.
大肠杆菌用 pCDF-1b-Pt NTT2 新鲜转化的 C41(DE3)16 在 2 × YT 中用链霉素生长过夜,然后稀释 (1:100) 到新鲜的 2 × YT 培养基中(每次摄取 1 ml 培养物与 [α-32P]-dATP;每次摄取 2 ml 培养物与 d5SICSTP 或 dNaMTP)补充有 50 mM 磷酸钾 (KPi) 和链霉素。具有无活性转运蛋白 pET-16b-Rp NTT2 的阴性对照,除了使用氨苄青霉素代替链霉素外,处理相同。细胞生长至约 0.6 的 OD600,并通过添加 IPTG (1 mM) 诱导 NTT 表达。让培养物再生长一小时(最终 OD600 约为 1.2),然后使用改编自前一篇论文15 的方法直接测定摄取,如下所述。
Preparation of media fraction for unnatural nucleoside triphosphate analysis
制备用于非天然核苷三磷酸分析的培养基馏分
The experiment was initiated by the addition of either dNaMTP or d5SICSTP (10 mM each) directly to the media to a final concentration of 0.25 mM. Cells were incubated with the substrate with shaking at 37 °C for 30 min and then pelleted (8,000 r.c.f. (relative centrifugal force) for 5 min, 4 °C). An aliquot of the media fraction (40 µl) was mixed with acetonitrile (80 µl) to precipitate proteins31, and then incubated at 22 °C for 30 min. Samples were either analysed immediately by HPLC or stored at −80 °C until analysis. Analysis began with centrifugation (12,000 r.c.f. for 10 min at 22 °C), then the pellet was discarded, and the supernatant was reduced to approximately 20 µl by SpeedVac, resuspended in buffer A (see below) to a final volume of 50 µl, and analysed by HPLC (see below).
通过将 dNaMTP 或 d5SICSTP(各 10 mM)直接添加到培养基中至终浓度为 0.25 mM 来开始实验。将细胞与底物在 37 °C 下振荡孵育 30 分钟,然后沉淀(8,000 r.c.f.(相对离心力)5 分钟,4 °C)。将等分试样的培养基组分 (40 μl) 与乙腈 (80 μl) 混合以沉淀蛋白质31,然后在 22 °C 下孵育 30 分钟。样品要么立即通过 HPLC 分析,要么储存在 −80 °C 下直至分析。分析从离心开始(在 22 °C 下 12,000 r.c.f. 10 分钟),然后弃去沉淀,通过 SpeedVac 将上清液还原至约 20 μl,重悬于缓冲液 A 中(见下文)至终体积为 50 μl,并通过 HPLC 分析(见下文)。
Preparation of cytoplasmic fraction for nucleoside triphosphate analysis
制备用于三磷酸核苷分析的细胞质组分
To analyse the intracellular desphosphorylation of the unnatural nucleoside triphosphate, cell pellets were subjected to 3 × 100 µl washes of ice-cold KPi (50 mM). Pellets were then resuspended in 250 µl of ice cold KPi (50 mM) and lysed with 250 µl of lysis buffer L7 of the PureLink Quick Plasmid DNA Miniprep Kit (200 mM NaOH, 1% w/v SDS), after which the resulting solution was incubated at 22 °C for 5 min. Precipitation buffer N4 (350 µl, 3.1 M potassium acetate, pH 5.5) was added, and the sample was mixed to homogeneity. Following centrifugation (>12,000 r.c.f. for 10 min, at 22 °C) the supernatant containing the unnatural nucleotides was applied to a Hypersep C18 solid phase extraction column (Thermo Scientific) prewashed with acetonitrile (1 ml) and buffer A (1 ml, see HPLC protocol for buffer composition). The column was then washed with buffer A and nucleotides were eluted with 1 ml of 50% acetonitrile:50% triethylammonium bicarbonate (TEAB) 0.1 M (pH 7.5). The eluent was reduced to approximately 50 µl in a SpeedVac and its volume was adjusted to 100 µl with buffer A before HPLC analysis.
为了分析非天然核苷三磷酸的细胞内去磷酸化,将细胞沉淀物用冰冷的 KPi (50 mM) 洗涤 3 × 100 μl。然后将沉淀重悬于 250 μl 冰冷的 KPi (50 mM) 中,并用 250 μl PureLink 快速质粒 DNA 小量制备试剂盒(200 mM NaOH,1% w/v SDS)的裂解缓冲液 L7 裂解,然后将所得溶液在 22 °C 下孵育 5 分钟。加入沉淀缓冲液 N4(350 μl,3.1 M 乙酸钾,pH 5.5),并将样品混合至均匀。离心后(>12,000 r.c.f.,22 °C,10 min),将含有非天然核苷酸的上清液加入用乙腈 (1 ml) 和缓冲液 A(1 ml,参见 HPLC 方案了解缓冲液组成)预洗的 Hypersep C18 固相萃取柱 (Thermo Scientific) 中。然后用缓冲液 A 洗涤色谱柱,用 1 ml 50% 乙腈:50% 碳酸氢三铵 (TEAB) 0.1 M (pH 7.5) 洗脱核苷酸。在 SpeedVac 中将洗脱液减少至约 50 μl,并在 HPLC 分析前用缓冲液 A 将其体积调节至 100 μl。
HPLC protocol and nucleoside triphosphate quantification
HPLC 方案和三磷酸核苷定量
Samples were applied to a Phenomenex Jupiter LC column (3 µm C18 300 Å, 250 × 4.6 mm) and subjected to a linear gradient of 0–40% B over 40 min at a flow rate of 1 ml min−1. Buffer A: 95% 0.1 M TEAB, pH 7.5; 5% acetonitrile. Buffer B: 20% 0.1 M TEAB, pH 7.5; 80% acetonitrile. Absorption was monitored at 230, 273, 288, 326 and 365 nm.
将样品上样到 Phenomenex Jupiter 液相色谱柱(3 μm C18 300 Å,250 × 4.6 mm)上,并以 1 ml min-1 的流速在 40 分钟内以 0–40% B 的线性梯度进行。缓冲液 A:95% 0.1 M TEAB,pH 7.5;5% 乙腈。缓冲液 B:20% 0.1 M TEAB,pH 7.5;80% 乙腈。在 230、273、288、326 和 365 nm 处监测吸收。
Each injection series included two extra control samples containing 5 nmol of dNaMTP or d5SICSTP. The areas under the peaks that corresponded to triphosphate, diphosphate, monophosphate and free nucleoside (confirmed by MALDI-TOF) were integrated for both the control and the unknown samples (described above). After peak integration, the ratio of the unknown peak to the control peak adjusted for the loss from the extraction step (62% and 70% loss for dNaM and d5SICS, respectively, Extended Data Table 3), provided a measure of the amount of each of the moieties in the sample. To determine the relative concentrations of unnatural nucleotide inside the cell, the amount of imported unnatural nucleotide (dXTP, µmol) was then divided by the volume of cells, which was calculated as the product of the volume of a single E. coli cell (1 µm3 based on a reported average value32; that is, 1 × 10−9 µl per cell) and the number of cells in each culture (OD600 of 1.0 equal to 1 × 109 cells per ml (ref. 32)). The RpNTT2 sample was used as a negative control and its signal was subtracted to account for incomplete washing of nucleotide species from the media.
每个进样系列包括两个额外的对照样品,含有 5 nmol 的 dNaMTP 或 d5SICSTP。对照样品和未知样品(如上所述)对对应于三磷酸盐、二磷酸盐、单磷酸盐和游离核苷(经 MALDI-TOF 证实)的峰下区域进行了积分。峰积分后,未知峰与对照峰的比率根据提取步骤的损失进行调整(dNaM 和 d5SICS 分别为 62% 和 70%,扩展数据表 3),提供了样品中每个部分的量度。为了确定细胞内非天然核苷酸的相对浓度,然后将输入的非天然核苷酸的量 (dXTP, μmol) 除以细胞体积,该体积计算为单个大肠杆菌细胞体积的乘积(1 μm3 基于报告的平均值32;即 每个细胞 1 × 10-9 μl)和每种培养物中的细胞数(OD600 为 1.0 等于 1 ×10 9 个细胞/ml(参考文献 32))。RpNTT2 样品用作阴性对照,并减去其信号以解释从培养基中核苷酸种类的不完全洗涤。
dATP uptake dATP 摄取
To analyse the intracellular desphosphorylation of dATP, after induction of the transporter, the uptake reaction was initiated by the addition of dATP (spiked with [α-32P]-dATP) to a final concentration of 0.25 mM, followed by incubation at 37 °C with shaking for 30 min. The culture was then centrifuged (8,000 r.c.f. for 5 min at 22 °C). Supernatant was analysed by TLC. Cell pellets were washed three times with ice-cold KPi (50 mM, 100 µl) to remove excess radioactive substrate, lysed with NaOH (0.2 M, 100 µl) and centrifuged (10,000 r.c.f. for 5 min at 22 °C) to remove cell debris; supernatant was analysed by TLC.
为了分析 dATP 的细胞内去磷酸化,在转运蛋白诱导后,通过添加 dATP(加标 [α-32P]-dATP)至终浓度为 0.25 mM 来启动摄取反应,然后在 37 °C 下振荡孵育 30 分钟。然后将培养物离心(8,000 r.c.f.,22 °C 下 5 分钟)。TLC 分析上清液。用冰冷的 KPi(50 mM,100 μl)洗涤细胞沉淀 3 次以去除多余的放射性底物,用 NaOH(0.2 M,100 μl)裂解并离心(10,000 r.c.f.,22 °C 下 5 分钟)以去除细胞碎片;TLC 分析上清液。
TLC analysis TLC 分析
Samples (1 µl) were applied on a 0.5 mm polyethyleneimine cellulose TLC plate and developed with sodium formate pH 3.0 (0.5 M, 30 s; 2.5 M, 2.5 min; 4.0 M, 40 min). Plates were dried using a heat gun and quantified by phosphorimaging (Storm Imager, Molecular Dynamics) and Quantity One software.
将样品 (1 μl) 施加在 0.5 mm 聚乙烯亚胺纤维素 TLC 板上,并用甲酸钠 pH 3.0 显影(0.5 M,30 秒;2.5 M,2.5 分钟;4.0 M,40 分钟)。使用热风枪干燥板,并通过磷光成像(Storm Imager、Molecular Dynamics)和 Quantity One 软件进行定量。
Optimization of nucleotide extraction from cells for HPLC injection
优化 HPLC 注射细胞中的核苷酸提取
To minimize the effect of the lysis and triphosphate extraction protocols on the decomposition of nucleoside triphosphate within the cell, the extraction procedure was optimized for the highest recovery with the lowest extent of decomposition (Extended Data Table 3). To test different extraction methods, cells were grown as described above, washed, and then 5 nmol of either dNaMTP or d5SICSTP was added to the pellets, which were then subjected to different extraction protocols including boiling water, hot ethanol, cold methanol, freeze and thaw, lysozyme, glass beads, NaOH, trichloroacetic acid (TCA) with Freon, and perchloric acid (PCA) with KOH33. The recovery and composition of the control was quantified by HPLC as described above to determine the most effective procedure. Method 3—that is, cell lysis with NaOH (Extended Data Table 3)—was found to be most effective and reproducible, thus we further optimized it by resuspension of the pellets in ice-cold KPi (50 mM, 250 µl) before addition of NaOH to decrease dephosphorylation after cell lysis (Method 4). Cell pellets were then processed as described above. See above for the final extraction protocol.
为了最大限度地减少裂解和三磷酸盐提取方案对细胞内三磷酸核苷分解的影响,对提取程序进行了优化,以实现最高的回收率和最低的分解程度(扩展数据表 3)。为了测试不同的提取方法,如上所述培养细胞,洗涤,然后将 5 nmol 的 dNaMTP 或 d5SICSTP 添加到沉淀中,然后进行不同的提取方案,包括沸水、热乙醇、冷甲醇、冻融、溶菌酶、玻璃珠、NaOH、含氟利昂的三氯乙酸 (TCA) 和含 KOH33 的高氯酸 (PCA).如上所述,通过 HPLC 定量对照的回收率和组成,以确定最有效的程序。方法 3 — 即用 NaOH 进行细胞裂解 (扩展数据表 3) — 被发现最有效且可重复,因此我们通过在添加 NaOH 之前将沉淀重悬于冰冷的 KPi (50 mM, 250 μl) 中来进一步优化它,以减少细胞裂解后的去磷酸化(方法 4)。然后按上述方法处理细胞沉淀。有关最终提取方案,请参见上文。
Preparation of the unnatural insert for pINF construction
用于 pINF 构建的非天然插入片段的制备
The TK-1-dNaM oligonucleotide containing dNaM was prepared using solid-phase DNA synthesis with ultra-mild DNA synthesis phosphoramidites on CPG ultramild supports (1 μmol, Glen Research, Sterling, Virginia, USA) and an ABI Expedite 8905 synthesizer. After the synthesis, the DMT-ON oligonucleotide was cleaved from the solid support, deprotected and purified by Glen-Pak cartridge according to the manufacturer's recommendation (Glen Research), and then subjected to 8 M urea 8% PAGE. The gel was visualized by ultraviolet shadowing, the band corresponding to the 75-mer was excised, and the DNA was recovered by crush and soak extraction, filtration (0.45 μm), and final desalting over Sephadex G-25 (NAP-25 Columns, GE Healthcare). The concentration of the single stranded oligonucleotide was determined by ultraviolet absorption at 260 nm assuming that the extinction coefficient of dNaM at 260 nm is equal to that of dA. TK-1-dNaM (4 ng) was next amplified by PCR under the following conditions: 1 × OneTaq reaction buffer, MgSO4 adjusted to 3.0 mM, 0.2 mM of dNTP, 0.1 mM of dNaMTP, 0.1 mM of the d5SICSTP analogue dTPT3TP, 1 μM of each of the primers pUC19-fusion-forward and pUC19-fusion-reverse, and 0.02 U μl−1 of OneTaq DNA Polymerase (in a total of 4 × 50 µl reactions) under the following thermal cycling conditions: initial denaturation (96 °C, 1 min) followed by 12 cycles of denaturation (96 °C, 10 s), annealing (60 °C, 15 s), and extension (68 °C, 2 min). An identical PCR without the unnatural triphosphates was run to obtain fully natural insert under identical conditions for the construction of the natural control plasmid. Reactions were subjected to spin column purification and then the desired PCR product (122 bp) was purified by a 4% agarose gel.
使用 CPG 超温和载体(1 μmol,Glen Research,Sterling,Virginia,USA)和 ABI Expedite 8905 合成仪上,使用固相 DNA 合成和超温和 DNA 合成亚磷酰胺制备含有 dNaM 的 TK-1-dNaM 寡核苷酸。合成后,将 DMT-ON 寡核苷酸从固体支持物中切割下来,根据制造商的建议 (Glen Research) 用 Glen-Pak 小柱脱保护和纯化,然后进行 8 M 尿素 8% PAGE 处理。通过紫外线阴影观察凝胶,切除对应于 75 聚体的条带,并通过粉碎和浸泡提取、过滤 (0.45 μm) 和 Sephadex G-25(NAP-25 色谱柱,GE Healthcare)的最终脱盐回收 DNA。假设 dNaM 在 260 nm 处的消光系数等于 dA 的消光系数,则通过 260 nm 处的紫外线吸收测定单链寡核苷酸的浓度。接下来在以下条件下通过 PCR 扩增 TK-1-dNaM (4 ng):1 × OneTaq 反应缓冲液,MgSO4 调节至 3.0 mM,0.2 mM dNTP,0.1 mM dNaMTP,0.1 mM d5SICSTP 类似物 dTPT3TP,1 μM 引物 pUC19 融合正向和 pUC19 融合反向,以及 0.02 U μl-1 OneTaq DNA 聚合酶(共 4 × 50 μl 反应),在以下热循环条件下: 初始变性(96°C,1分钟),然后是12个变性循环(96°C,10秒),退火(60°C,15秒)和延伸(68°C,2分钟)。运行不含非天然三磷酸盐的相同 PCR,在相同条件下获得完全天然插入片段,用于构建天然对照质粒。对反应进行离心柱纯化,然后用 4% 琼脂糖凝胶纯化所需的 PCR 产物 (122 bp)。
pUC19 linearization for pINF construction
用于 pINF 构建的 pUC19 线性化
pUC19 (20 ng) was amplified by PCR under the following conditions: 1 × Q5 reaction buffer, MgSO4 adjusted to 3.0 mM, 0.2 mM of dNTP, 1 μM of each primers pUC19-lin-forward and pUC19-lin-reverse, and 0.02 U μl−1 of Q5 Hot Start High-Fidelity DNA Polymerase (in a total of 4 × 50 µl reactions with one reaction containing 0.5 × Sybr Green I) under the following thermal cycling conditions: initial denaturation (98 °C, 30 s); 20 cycles of denaturation (98 °C, 10 s), annealing (60 °C, 15 s), and extension (72 °C, 2 min); and final extension (72 °C, 5 min). The desired PCR product (2,611 bp) was purified by a 2% agarose gel.
在以下条件下通过 PCR 扩增 pUC19 (20 ng):1 × Q5 反应缓冲液,MgSO4 调节至 3.0 mM,0.2 mM dNTP,pUC19 lin-正向和 pUC19-lin-反向引物 1 μM,以及 0.02 U μl-1 Q5 热启动高保真 DNA 聚合酶(总共 4 × 50 μl 反应,其中 1 次反应含有 0.5 × Sybr Green I): 初始变性(98 °C,30 s);20 个变性(98 °C,10 秒),退火(60 °C,15 秒)和延伸(72 °C,2 分钟)循环;和最终延伸(72 °C,5 分钟)。所需的 PCR 产物 (2,611 bp) 用 2% 琼脂糖凝胶纯化。
PCR assembly of pINF and the natural control plasmid
pINF 和天然对照质粒的 PCR 组装
A linear fragment was amplified from pUC19 using primers pUC19-lin-forward and pUC19-lin-reverse. The resulting product (800 ng, 4.6 × 10−13 mol) was combined with either the natural or unnatural insert (see above) (56 ng, 7.0 × 10−13 mol) and assembled by circular overlap extension PCR under the following conditions: 1 × OneTaq reaction buffer, MgSO4 adjusted to 3.0 mM, 0.2 mM of dNTP, 0.1 mM of dNaMTP, 0.1 mM of the d5SICSTP analogue dTPT3TP, and 0.02 U μl−1 of OneTaq DNA Polymerase (in a total of 4 × 50 µl reactions with one reaction containing 0.5 × Sybr Green I) using the following thermal cycling conditions: initial denaturation (96 °C, 1 min); 12 cycles of denaturation (96 °C, 30 s), annealing (62 °C, 1 min), and extension (68 °C, 5 min); final extension (68 °C, 5 min); and slow cooling (68 °C to 10 °C at a rate of −0.1 °C s−1). The PCR product was analysed by restriction digestion on 1% agarose and used directly for E. coli transformation. The d5SICS analogue dTPT322 pairs with dNaM, and dTPT3TP was used in place of d5SICSTP as DNA containing dTPT3–dNaM is better PCR amplified than DNA containing d5SICS–dNaM, and this allowed for differentiation of synthetic and in vivo replicated pINF, as well as facilitated the construction of high-quality pINF (UBP content >99%).
使用引物 pUC19-lin-forward 和 pUC19-lin-reverse 从 pUC19 扩增线性片段。将所得产物(800 ng,4.6 × 10-13 mol)与天然或非天然插入片段(见上文)(56 ng,7.0 × 10-13 mol)混合,并在以下条件下通过环重叠延伸 PCR 组装:1 × OneTaq 反应缓冲液,MgSO4 调节至 3.0 mM,0.2 mM dNTP,0.1 mM dNaMTP,0.1 mM d5SICSTP 类似物 dTPT3TP, 和 0.02 U μl-1 OneTaq DNA 聚合酶(总共 4 × 50 μl 反应,其中 1 次反应含有 0.5 × Sybr Green I):初始变性(96 °C,1 分钟);12 个变性(96 °C,30 秒),退火(62 °C,1 分钟)和延伸(68 °C,5 分钟)循环;最终延伸(68 °C,5 分钟);和缓慢冷却(68 °C 至 10 °C,速率为 -0.1 °C s-1)。通过对 1% 琼脂糖进行限制性消化分析 PCR 产物,并直接用于大肠杆菌转化。d5SICS 类似物 dTPT322 对与 dNaM 和 dTPT3TP 代替 d5SICSTP,因为含有 dTPT3-dNaM 的 DNA 比含有 d5SICS-dNaM 的 DNA 扩增效果更好,这允许区分合成和体内复制的 pINF,并促进高质量 pINF 的构建(UBP 含量 >99%)。
Preparation of electrocompetent cells for pINF replication in E. coli
用于大肠杆菌中 pINF 复制的电感受态细胞的制备
C41(DE3) cells were transformed by heat shock34 with 200 ng of pACS plasmid, and the transformants were selected overnight on 2 × YT-agar supplemented with streptomycin. A single clone of freshly transformed C41(DE3) pACS was grown overnight in 2 × YT medium (3 ml) supplemented with streptomycin and KPi (50 mM). After 100-fold dilution into the same fresh 2 × YT media (300 ml), the cells were grown at 37 °C until they reached an OD600 of 0.20 at which time IPTG was added to a final concentration of 1 mM to induce the expression of PtNTT2. Cells were grown for another 40 min and then growth was stopped by rapid cooling in ice water with intensive shaking. After centrifugation in a prechilled centrifuge (2,400 r.c.f. for 10 min, 4 °C), the spent media was removed, and the cells were prepared for electroporation by washing with ice-cold sterile water (3 × 150 ml). After washing, the cells were resuspended in ice-cold 10% glycerol (1.5 ml) and split into 50-µl aliquots. Although we found that dry ice yielded better results than liquid nitrogen for freezing cells to store for later use, freshly prepared cells were used for all reported experiments as they provided higher transformation efficiency of pINF and higher replication fidelity of the UBP.
用 200 ng pACS 质粒热休克34 转化 C41(DE3) 细胞,并在 2 × 补充链霉素的 YT-琼脂上过夜选择转化体。新鲜转化的 C41 (DE3) pACS 的单个克隆在补充有链霉素和 KPi (50 mM) 的 2 × YT 培养基 (3 ml) 中生长过夜。将细胞稀释到相同的新鲜 2 × YT 培养基 (300 ml) 中 100 倍后,将细胞在 37 °C 下生长直至达到 OD600 的 0.20,此时加入 IPTG 至终浓度为 1 mM 以诱导 PtNTT2 的表达。将细胞再生长 40 分钟,然后在冰水中快速冷却并强烈摇动来停止生长。在预冷的离心机(2,400 r.c.f.,10 min,4 °C)中离心后,除去用过的培养基,并通过用冰冷的无菌水(3 × 150 ml)洗涤来制备细胞以进行电穿孔。洗涤后,将细胞重悬于冰冷的 10% 甘油 (1.5 ml) 中,并分成 50 μl 等分试样。尽管我们发现干冰在冷冻细胞以备后用方面比液氮产生更好的结果,但所有报道的实验都使用新鲜制备的细胞,因为它们提供了更高的 pINF 转化效率和更高的 UBP 复制保真度。
Electroporation and recovery for pINF replication in E. coli
电穿孔和回收大肠杆菌中 pINF 复制
The aliquot of cells was mixed with 2 µl of plasmid (400 ng), transferred to 0.2 cm gap electroporation cuvette and electroporated using a Bio-Rad Gene Pulser according to the manufacturer’s recommendations (voltage 25 kV, capacitor 2.5 µF, resistor 200 Ω, time constant 4.8 ms). Pre-warmed 2 × YT media (0.95 ml, streptomycin, 1 mM IPTG, 50 mM KPi) was added, and after mixing, 45 µl was removed and combined with 105 µl of the same media (3.33-fold dilution) supplemented with 0.25 mM of dNaMTP and d5SICSTP. The resulting mixture was allowed to recover for 1 h at 37 °C with shaking (210 revolutions per min (r.p.m.)). The original transformation media (10 µl) was spread onto 2 × YT-agar containing streptomycin with 10- and 50-fold dilutions for the determination of viable colony forming units after overnight growth at 37 °C to calculate the number of the transformed pINF molecules (see the section on calculation of the plasmid amplification). Transformation, recovery and growth were carried out identically for the natural control plasmid. In addition, a negative control was run and treated identically to pINF transformation except that it was not subjected to electroporation (Extended Data Fig. 7b). No growth in the untransformed negative control samples was observed even after 6 days. No PCR amplification of the negative control was detected, which confirms that unamplified pINF plasmid is not carried through cell growth and later detected erroneously as the propagated plasmid.
将细胞等分试样与 2 μl 质粒 (400 ng) 混合,转移至 0.2 cm 间隙电穿孔比色皿中,并根据制造商的建议(电压 25 kV,电容器 2.5 μF,电阻器 200 Ω,时间常数 4.8 ms)使用 Bio-Rad 基因脉冲器进行电穿孔。加入预热的 2 × YT 培养基(0.95 ml、链霉素、1 mM IPTG、50 mM KPi),混合后,除去 45 μl 并与 105 μl 补充有 0.25 mM dNaMTP 和 d5SICSTP 的相同培养基(3.33 倍稀释)混合。将所得混合物在 37 °C 下振荡 (210 转/分钟 (r.p.m.)) 恢复 1 小时。将原始转化培养基 (10 μl) 涂布在 2 ×含有 10 倍和 50 倍稀释链霉素的 YT-琼脂上,用于在 37 °C 下过夜生长后测定活菌落形成单位,以计算转化的 pINF 分子的数量(参见质粒扩增的计算部分)。天然对照质粒的转化、恢复和生长过程相同。此外,阴性对照的运行和处理与 pINF 转化相同,只是它没有进行电穿孔(扩展数据图 7b)。即使在 6 天后,未转化的阴性对照样品也没有观察到生长。未检测到阴性对照的 PCR 扩增,这证实了未扩增的 pINF 质粒未通过细胞生长携带,后来被错误地检测为繁殖的质粒。
Analysis of pINF replication in E. coli
大肠杆菌中 pINF 复制的分析
After recovery, the cells were centrifuged (4,000 r.c.f. for 5 min, 4 °C), and spent media (0.15 ml) was removed and analysed for nucleotide composition by HPLC (Extended Data Fig. 7a). The cells were resuspended in fresh 2 × YT media (1.5 ml, streptomycin, ampicillin, 1 mM IPTG, 50 mM KPi, 0.25 mM dNaMTP, 0.25 mM d5SICSTP) and grown overnight at 37 °C while shaking (250 r.p.m.), resulting in tenfold dilution compared to recovery media or 33.3-fold dilution compared to the originally transformed cells. Aliquots (100 µl) were taken after 15, 19, 24, 32, 43, 53, 77 and 146 h, OD600 was determined, and the cells were centrifuged (8,000 r.c.f. for 5 min, 4 °C). Spent media were analysed for nucleotide composition by HPLC (Extended Data Fig. 7a), and the pINF and pACS plasmid mixtures were recovered and linearized with NdeI restriction endonuclease; pINF plasmid was purified by 1% agarose gel electrophoresis (Extended Data Fig. 7b) and analysed by LC-MS/MS. The retention of the UBP on the pINF plasmid was quantified by biotin gel shift mobility assay and sequencing as described below.
回收后,将细胞离心(4,000 r.c.f.,5 分钟,4 °C),除去用过的培养基 (0.15 ml),并通过 HPLC 分析核苷酸组成(扩展数据图 7a)。将细胞重悬于新鲜的 2 × YT 培养基(1.5 ml、链霉素、氨苄青霉素、1 mM IPTG、50 mM KPi、0.25 mM dNaMTP、0.25 mM d5SICSTP)中,并在 37 °C 下摇动 (250 rpm) 生长过夜,与回收培养基相比稀释 10 倍,与原始转化细胞相比稀释 33.3 倍。15、19、24、32、43、53、77 和 146 小时后取等分试样 (100 μl),测定 OD600,并将细胞离心(8,000 r.c.f.,5 分钟,4 °C)。通过 HPLC 分析用过的培养基的核苷酸组成(扩展数据图 7a),回收 pINF 和 pACS 质粒混合物并用 NdeI 限制性核酸内切酶线性化;通过 1% 琼脂糖凝胶电泳纯化 pINF 质粒(扩展数据图 7b),并通过 LC-MS/MS 分析。如下所述,通过生物素凝胶迁移率测定和测序定量 UBP 在 pINF 质粒上的保留。
Mass spectrometry of pINF
pINF 的质谱分析
Linearized pINF was digested to nucleosides by treatment with a mixture of nuclease P1 (Sigma-Aldrich), shrimp alkaline phosphatase (NEB), and DNase I (NEB), overnight at 37 °C, following a previously reported protocol25. LC-MS/MS analysis was performed in duplicate by injecting 15 ng of digested DNA on an Agilent 1290 UHPLC equipped with a G4212A diode array detector and a 6490A Triple Quadrupole Mass Detector operating in the positive electrospray ionization mode (+ESI). UHPLC was carried out using a Waters XSelect HSS T3 XP column (2.1 × 100 mm, 2.5 µm) with the gradient mobile phase consisting of methanol and 10 mM aqueous ammonium formate (pH 4.4). MS data acquisition was performed in Dynamic Multiple Reaction Monitoring (DMRM) mode. Each nucleoside was identified in the extracted chromatogram associated with its specific MS/MS transition: dA at m/z 252→136, d5SICS at m/z 292→176, and dNaM at m/z 275→171. External calibration curves with known amounts of the natural and unnatural nucleosides were used to calculate the ratios of individual nucleosides within the samples analysed. LC-MS/MS quantification was validated using synthetic oligonucleotides1 containing unnatural d5SICS and dNaM (Extended Data Table 2).
按照先前报道的方案25,用核酸酶 P1 (Sigma-Aldrich)、虾碱性磷酸酶 (NEB) 和 DNase I (NEB) 的混合物在 37 °C 下过夜,将线性化的 pINF 消化成核苷。在配备 G4212A 二极管阵列检测器和 6490A 三重四极杆质谱检测器的 Agilent 1290 UHPLC 上注入 15 ng 消化的 DNA,在正电喷雾电离模式 (+ESI) 下运行,进行一式两份的 LC-MS/MS 分析。使用Waters XSelect HSS T3 XP色谱柱(2.1 × 100 mm,2.5 μm)和由甲醇和10 mM甲酸铵水溶液(pH 4.4)组成的梯度流动相进行UHPLC。在动态多反应监测 (DMRM) 模式下进行 MS 数据采集。在提取的色谱图中鉴定出与其特异性 MS/MS 通道相关的每种核苷:dA 在 m/z 252→136 处,d5SICS 在 m/z 292→176 处,dNaM 在 m/z 275→171 处。使用已知量天然和非天然核苷的外部校准曲线计算分析样品中单个核苷的比率。使用含有非天然 d5SICS 和 dNaM 的合成寡核苷酸1 验证 LC-MS/MS 定量(扩展数据表 2)。
DNA biotinylation by PCR to measure fidelity by gel shift mobility assay
通过 PCR 进行 DNA 生物素化,通过凝胶位移迁移率测定法测量保真度
Purified mixtures of pINF and pACS plasmids (1 ng) from growth experiments were amplified by PCR under the following conditions: 1 × OneTaq reaction buffer, MgSO4 adjusted to 3.0 mM, 0.3 mM of dNTP, 0.1 mM of the biotinylated dNaMTP analogue dMMO2SSBIOTP, 0.1 mM of d5SICSTP, 1 μM of each of the primers pUC19-seq-forward and pUC19-seq-reverse, 0.02 U μl−1 of OneTaq DNA Polymerase, and 0.0025 U μl−1 of DeepVent DNA Polymerase in a total volume of 25 μl in an CFX Connect Real-Time PCR Detection System (Bio-Rad) under the following thermal cycling conditions: initial denaturation (96 °C, 1 min); 10 cycles of denaturation (96 °C, 30 s), annealing (64 °C, 30 s), and extension (68 °C, 4 min). PCR products were purified, and the resulting biotinylated DNA duplexes (5 μl, 25–50 ng) were mixed with streptavidin (1 μl, 1 μg μl−1, Promega) in phosphate buffer (50 mM sodium phosphate, pH 7.5, 150 mM NaCl, 1 mM EDTA), incubated for 30 min at 37 °C, mixed with 5 × non-denaturing loading buffer (Qiagen), and loaded onto 6% non-denaturing PAGE. After running at 110 V for 30 min, the gel was visualized and quantified. The resulting fragment (194 bp) with primer regions underlined and the unnatural nucleotide in bold (X represents dNaM or its biotinylated analogue dMMO2SSBIO) is 5′-GCAGGCATGCAAGCTTGGCGTAATCATGG TCATAGCTGTTTCCTGTGTGAAATTGTTATCCGCTCACAXTTCCACACAACATACGAGCCGGA AGCATAAAGTGTAAAGCCTGGGGTGCCTAATGAGTGAGCTAACTCACATTAATTGCGTTGCGCTCACTGCCCGCTTT CCAGTCGGGAAACCTGTCGTGCCAG.
在以下条件下,通过 PCR 扩增来自生长实验的 pINF 和 pACS 质粒 (1 ng) 的纯化混合物:1 × OneTaq 反应缓冲液,MgSO4 调节至 3.0 mM,0.3 mM dNTP,0.1 mM 生物素化 dNaMTP 类似物 dMMO2SSBIOTP,0.1 mM d5SICSTP,1 μM 引物 pUC19-seq-forward 和 pUC19-seq-reverse 各 1 μM, 在以下热循环条件下,在 CFX Connect 实时 PCR 检测系统 (Bio-Rad) 中,总体积为 25 μl 的 0.02 U μl-1 OneTaq DNA 聚合酶和 0.0025 U μl-1 的 DeepVent DNA 聚合酶:初始变性(96 °C,1 分钟);10 个变性(96 °C,30 秒)、退火(64 °C,30 秒)和延伸(68 °C,4 分钟)循环。纯化 PCR 产物,将得到的生物素化 DNA 双链体(5 μl,25-50 ng)与链霉亲和素(1 μl,1 μg μl-1,Promega)在磷酸盐缓冲液(50 mM 磷酸钠,pH 7.5,150 mM NaCl,1 mM EDTA)中混合,在 37 °C 下孵育 30 分钟,与 5 ×非变性上样缓冲液 (Qiagen) 混合,并加载到 6% 非变性 PAGE 上。在 110 V 下运行 30 分钟后,对凝胶进行可视化和定量。所得片段 (194 bp) 带有引物区域下划线,非天然核苷酸以粗体显示(X 代表 dNaM 或其生物素化类似物 dMMO2SSBIO)是 5′-GCAGGCATGCAAGCAAGCTTGGCGTAATCATGG TCATAGCTGTTTCCTGTGTGAAATTGTTATCCGCTCACAXTTCCACACAACATACACACGAGCCGGA AGCATAAAGTGTAAAGCCTGGGGTGCCTAACTCACATTAATTGCGTTGCGCTCACTGCCCGCTTT CCAGTCGGGAAACCTGTCGTGCCAG。
Streptavidin shift calibration for gel shift mobility assay
用于凝胶迁移率测定的链霉亲和素迁移校准
We have already reported a calibration between streptavidin shift and the fraction of sequences with UBP in the population (see Supplementary Fig. 8 of ref. 1). However, we found that spiking the PCR reaction with DeepVent improves the fidelity with which DNA containing d5SICS-dMMO2SSBIO is amplified, and thus we repeated the calibration with added DeepVent. To quantify the net retention of the UBP, nine defined mixtures of the TK-1-dNaM template and its fully natural counterpart were prepared (Extended Data Fig. 6a), subjected to biotinylation by PCR and analysed by mobility-shift assay on 6% non-denaturing PAGE as described above. For calibration, the mixtures TK-1-dNaM template and its fully natural counterpart with a known ratio of unnatural and natural templates (0.04 ng) were amplified under the same conditions over nine cycles of PCR with pUC19-fusion primers and analysed identically to samples from the growth experiment (see the section on DNA biotinylation by PCR). Each experiment was run in triplicate (a representative gel assay is shown in Extended Data Fig. 6b), and the streptavidin shift (SAS, %) was plotted as function of the UBP content (UBP, %). The data was then fit to a linear equation, SAS = 0.77 × UBP + 2.0 (R2 = 0.999), where UBP corresponds to the retention of the UBP (%) in the analysed samples after cellular replication and was calculated from the SAS shift using the equation above.
我们已经报道了链霉亲和素转移与群体中具有 UBP 的序列分数之间的校准(参见参考文献 1 的补充图 8)。然而,我们发现用 DeepVent 加标 PCR 反应可以提高含有 d5SICS-dMMO2SSBIO 的 DNA 扩增的保真度,因此我们使用添加的 DeepVent 重复校准。为了量化 UBP 的净保留,制备了 TK-1-dNaM 模板及其完全天然对应物的 9 种确定混合物(扩展数据图 6a),通过 PCR 进行生物素化,并通过 6% 非变性 PAGE 的迁移率变化测定进行分析如上所述。对于校准,在相同条件下,用 pUC19 融合引物在 9 个循环的 PCR 中扩增混合物 TK-1-dNaM 模板及其具有已知比例的非天然和天然模板 (0.04 ng) 的完全天然对应物,并与生长实验中的样品相同(参见 PCR 的 DNA 生物素化部分)。每个实验一式三份进行(代表性凝胶测定如扩展数据图 6b 所示),链霉亲和素位移 (SAS, %) 绘制为 UBP 含量 (UBP, %) 的函数。然后将数据拟合到线性方程 SAS = 0.77 × UBP + 2.0 (R2 = 0.999),其中 UBP 对应于细胞复制后分析样品中 UBP 的保留率 (%),并使用上述方程根据 SAS 偏移计算。
Calculation of plasmid amplification
质粒扩增的计算
The cells were plated on 2 × YT-agar containing ampicillin and streptavidin directly after transformation with pINF, and the colonies were counted after overnight growth at 37 °C. Assuming each cell is only transformed with one molecule of plasmid, colony counts correspond to the original amount of plasmid that was taken up by the cells. After overnight growth, the plasmids were purified from a specific volume of the cell culture and quantified. As purified plasmid DNA represents a mixture of the pINF and pACS plasmids, digestion restriction analysis with NdeI exonuclease was performed to linearize both plasmids, followed by 1% agarose gel electrophoresis (Extended Data Fig. 7b). An example of calculations for the 19-h time point with one of three triplicates is provided in Supplementary Information.
用 pINF 转化后,将细胞直接接种在含有氨苄青霉素和链霉亲和素的 2 × YT-琼脂上,并在 37 °C 下生长过夜后对集落进行计数。 假设每个细胞仅用一个质粒分子转化,则菌落计数对应于细胞吸收的原始质粒量。过夜生长后,从特定体积的细胞培养物中纯化质粒并定量。由于纯化的质粒 DNA 代表 pINF 和 pACS 质粒的混合物,因此使用 NdeI 核酸外切酶进行消化限制性分析以线性化两种质粒,然后进行 1% 琼脂糖凝胶电泳(扩展数据图 7b)。补充信息中提供了三个一式三份之一的 19 小时时间点的计算示例。
Fragment generation for Sanger sequencing to measure fidelity
用于 Sanger 测序的片段生成以测量保真度
Purified mixtures of pINF and pACS plasmids (1 ng) after the overnight growth were amplified by PCR under the following conditions: 1 × OneTaq reaction buffer, MgSO4 adjusted to 3.0 mM, 0.2 mM of dNTP, 0.1 mM of dNaMTP, 0.1 mM of the d5SICSTP analogue dTPT3TP, 1 μM of each of the primers pUC19-seq2-forward and pUC19-seq-reverse (see below), and 0.02 U μl−1 of OneTaq DNA Polymerase in a total volume of 25 μl under the following thermal cycling conditions: initial denaturation (96 °C, 1 min); and 10 cycles of denaturation (96 °C, 30 s), annealing (64 °C, 30 s), and extension (68 °C, 2 min). Products were purified by spin column, quantified to measure DNA concentration and then sequenced as described below. The sequenced fragment (304 bp) with primer regions underlined and the unnatural nucleotide in bold (X, dNaM) is 5′-GCTGCAAGGCGATTAAGTTGGGTAACGCC AGGGTTTTCCCAGTCACGACGTTGTAAAACGACGGCCAGTGAATTCGAGCTCGGTACCCGGG GATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGCGTAATCATGGTCATAGCTGTT TCCTGTGTGAAATTGTTATCCGCTCACAXTTCCACACAACATACGA GCCGGAAGCATAAAGTGTAAAGCCTGGGGTGCCTAATGAGTGAGCTAACTCACATTAATTGCGTTGCGC TCACTGCCCGCTTTCCAGTCGGGAAACCTGTCGTGCCAG.
在以下条件下,通过PCR扩增过夜生长后纯化的pINF和pACS质粒混合物(1 ng):1 × OneTaq 反应缓冲液,MgSO4 调节至 3.0 mM,0.2 mM dNTP,0.1 mM dNaMTP,0.1 mM d5SICSTP 类似物 dTPT3TP,1 μM 引物 pUC19-seq2-正向和 pUC19-seq-反向引物(见下文), 在以下热循环条件下,总体积为 25 μl 的 0.02 U μl-1 OneTaq DNA 聚合酶:初始变性(96 °C,1 分钟);和 10 个变性(96 °C,30 秒),退火(64 °C,30 秒)和延伸(68 °C,2 分钟)循环。通过离心柱纯化产物,定量以测量 DNA 浓度,然后如下所述进行测序。测序片段 (304 bp),引物区域下划线,非天然核苷酸以粗体 (X, dNaM) 显示 5′-GCTGCAAGGCGATTAAGTTGGGTAACGCC AGGGTTTTCCCAGTCACGACGTTGTAAAACGACGGCAGTGAATTCGAGCTCGGTCAGGAGGAGGGCAGAGGCAAGCTTGGCGTAATCATGGTCATAGCTGTT TCCTGTGTGAAATTGTTATCCGCTCTCAXTTCCACACAAGCATAAAGTAAAGCCTGGAGAGGTGAAGTAAGGAACTCATTAATTGCGTTGCGC TCACTGCCCTTTCCAGTCGGGAAACCTGTCGTGCCAG。
Sanger sequencing Sanger 测序
The cycle sequencing reactions (10 μl) were performed on a 9800 Fast Thermal Cycler (Applied Biosystems) with the Cycle Sequencing Mix (0.5 μl) of the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) containing 1 ng template and 6 pmol of sequencing primer pUC19-seq-reverse under the following thermal cycling conditions: initial denaturation (98 °C, 1 min); and 25 cycles of denaturation (96 °C, 10 s), annealing (60 °C, 15 s), and extension (68 °C, 2.5 min). Upon completion, the residual dye terminators were removed from the reaction with Agencourt CleanSEQ (Beckman-Coulter, Danvers, Massachusetts, USA). Products were eluted off the beads with deionized water and sequenced directly on a 3730 DNA Analyzer (Applied Biosystems). Sequencing traces were collected using Applied Biosystems Data Collection software v3.0 and analysed with the Applied Biosystems Sequencing Analysis v5.2 software.
在含有 1 ng 模板和 6 pmol 测序引物 pUC19-seq-reverse 的 BigDye Terminator v3.1 循环测序试剂盒 (Applied Biosystems) 的循环测序混合物 (0.5 μl) 的 9800 Fast 热循环仪 (Applied Biosystems) 上进行循环测序反应 (10 μl),在以下热循环条件下:初始变性(98 °C,1 分钟);和 25 个变性 (96 °C, 10 s) 、退火 (60 °C, 15 s) 和延伸 (68 °C, 2.5 分钟) 循环。完成后,使用 Agencourt CleanSEQ(美国马萨诸塞州丹佛斯市 Beckman-Coulter)从反应中去除残留的染料终止剂。用去离子水从磁珠上洗脱产物,并直接在 3730 DNA 分析仪 (Applied Biosystems) 上测序。使用 Applied Biosystems 数据收集软件 v3.0 收集测序痕迹,并使用 Applied Biosystems 测序分析 v5.2 软件进行分析。
Analysis of Sanger sequencing traces
Sanger 测序轨迹分析
Sanger sequencing traces were analysed as described previously1,26 to determine the retention of the unnatural base pair. In brief, the presence of an unnatural nucleotide leads to a sharp termination of the sequencing profile, whereas mutation to a natural nucleotide results in ‘read-through’. The extent of this read-through after normalization is inversely correlated with the retention of the unnatural base pair. Raw sequencing traces were analysed by first adjusting the start and stop points for the Sequencing Analysis software (Applied Biosystems) and then determining the average signal intensity individually for each channel (A, C, G and T) for peaks within the defined points. This was done separately for the parts of the sequencing trace before (section L) and after (section R) the unnatural nucleotide. The R/L ratio after normalization (R/L)norm for sequencing decay and read-through in the control unamplified sample (R/L = 0.55(R/L)norm + 7.2, see ref. 26 for details) corresponds to the percentage of the natural sequences in the pool. Therefore, an overall retention (F) of the incorporation of the unnatural base pair during PCR is equal to 1 – (R/L)norm. As significant read-through (over 20%) was observed in the direction of the pUC19-seq2-forward primer even with the control plasmid (synthetic pINF); sequencing of only the opposite direction (pUC19-seq-reverse) was used to gauge fidelity. Raw sequencing traces are shown in Fig. 2f and provided as Supplementary Data.
如前所述分析 Sanger 测序痕迹 1,26 以确定非自然碱基对的保留。简而言之,非天然核苷酸的存在会导致测序谱的急剧终止,而突变为天然核苷酸会导致“通读”。归一化后这种读取的程度与非自然碱基对的保留呈负相关。首先调整测序分析软件 (Applied Biosystems) 的起点和终点,然后分别确定定义点内峰的每个通道(A、C、G 和 T)的平均信号强度,从而分析原始测序痕迹。这是对非天然核苷酸之前(L 部分)和之后(R 部分)的测序轨迹部分分别进行的。对照未扩增样品中测序衰减和通读的归一化 (R/L) 规范后的 R/L 比率 (R/L = 0.55(R/L)norm + 7.2,详见参考文献 26)对应于池中自然序列的百分比。因此,在 PCR 过程中掺入非天然碱基对的总保留 (F) 等于 1 – (R/L)标准。因为即使使用对照质粒(合成 pINF),在 pUC19-seq2 正向引物的方向上也观察到显著的通读 (超过 20%);仅使用相反方向的测序 (pUC19-seq-reverse) 来衡量保真度。原始测序轨迹如图 2f 所示,并作为补充数据提供。
References 引用
Malyshev, D. A. et al. Efficient and sequence-independent replication of DNA containing a third base pair establishes a functional six-letter genetic alphabet. Proc. Natl Acad. Sci. USA 109, 12005–12010 (2012)
Malyshev, DA 等人。对包含第三个碱基对的 DNA 进行高效且不依赖于序列的复制,建立了一个功能性的六字母遗传字母表。美国国家科学院院刊 109, 12005–12010 (2012)Yang, Z., Chen, F., Alvarado, J. B. & Benner, S. A. Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J. Am. Chem. Soc. 133, 15105–15112 (2011)
Yang, Z., Chen, F., Alvarado, J. B. & Benner, S. A. 一个由六个字母组成的合成遗传系统的扩增、突变和测序。J. Am. Chem. Soc.133, 15105–15112 (2011)Yamashige, R. et al. Highly specific unnatural base pair systems as a third base pair for PCR amplification. Nucleic Acids Res. 40, 2793–2806 (2012)
Yamashige, R. 等人。高度特异性的非天然碱基对系统作为 PCR 扩增的第三个碱基对。核酸研究40, 2793–2806 (2012)Seo, Y. J., Malyshev, D. A., Lavergne, T., Ordoukhanian, P. & Romesberg, F. E. Site-specific labeling of DNA and RNA using an efficiently replicated and transcribed class of unnatural base pairs. J. Am. Chem. Soc. 133, 19878–19888 (2011)
Seo, Y. J., Malyshev, DA, Lavergne, T., Ordoukhanian, P. & Romesberg, FE 使用一类有效复制和转录的非自然碱基对DNA和RNA进行位点特异性标记。J. Am. Chem. Soc.133, 19878–19888 (2011)Seo, Y. J., Matsuda, S. & Romesberg, F. E. Transcription of an expanded genetic alphabet. J. Am. Chem. Soc. 131, 5046–5047 (2009)
Seo, Y. J., Matsuda, S. & Romesberg, F. E. 扩展遗传字母表的转录。J. Am. Chem. Soc.131, 5046–5047 (2009)Betz, K. et al. Structural insights into DNA replication without hydrogen bonds. J. Am. Chem. Soc. 135, 18637–18643 (2013)
Betz, K. 等人。对无氢键的 DNA 复制的结构见解。J. Am. Chem. Soc.135, 18637–18643 (2013)Betz, K. et al. KlenTaq polymerase replicates unnatural base pairs by inducing a Watson–Crick geometry. Nature Chem. Biol. 8, 612–614 (2012)
Betz, K. 等人。KlenTaq 聚合酶通过诱导 Watson-Crick 几何形状来复制非自然碱基对。Nature Chem. Biol.8, 612–614 (2012)Wu, Y., Fa, M., Tae, E. L., Schultz, P. G. & Romesberg, F. E. Enzymatic phosphorylation of unnatural nucleosides. J. Am. Chem. Soc. 124, 14626–14630 (2002)
Wu, Y., Fa, M., Tae, E. L., Schultz, P. G. & Romesberg, FE 非天然核苷的酶磷酸化。J. Am. Chem. Soc.124, 14626–14630 (2002 年)Yan, H. & Tsai, M. D. Nucleoside monophosphate kinases: structure, mechanism, and substrate specificity. Adv. Enzymol. 73, 103–134 (1999)
Yan, H. & Tsai, M. D. 核苷单磷酸激酶:结构、机制和底物特异性。Adv. Enzymol.73, 103–134 (1999)Winkler, H. H. & Neuhaus, H. E. Non-mitochondrial ATP transport. Trends Biochem. Sci. 24, 64–68 (1999)
Winkler, H. H. & Neuhaus, H. E. 非线粒体ATP运输。趋势 生物化学 科学24, 64–68 (1999 年)Amiri, H., Karlberg, O. & Andersson, S. G. Deep origin of plastid/parasite ATP/ADP translocases. J. Mol. Evol. 56, 137–150 (2003)
Amiri, H., Karlberg, O. & Andersson, SG. 质体/寄生虫ATP/ADP转位酶的深层起源。J. Mol. Evol.56, 137–150 (2003)Hatch, T. P., Al-Hossainy, E. & Silverman, J. A. Adenine nucleotide and lysine transport in Chlamydia psittaci. J. Bacteriol. 150, 662–670 (1982)
Hatch, T. P., Al-Hossainy, E. & Silverman, J. A. 腺嘌呤核苷酸和赖氨酸在鹦鹉热衣原体中的转运。J. 细菌。150, 662–670 (1982 年)Winkler, H. H. Rickettsial permeability: an ADP-ATP transport system. J. Biol. Chem. 251, 389–396 (1976)
Winkler, H. H. 立克次体通透性:ADP-ATP 运输系统。J. Biol. 化学。251, 389–396 (1976 年)Horn, M. & Wagner, M. Bacterial endosymbionts of free-living amoebae. J. Eukaryot. Microbiol. 51, 509–514 (2004)
Horn, M. & Wagner, M. 自由生活变形虫的细菌内共生体。J. 真核生物。微生物学。51, 509–514 (2004)Haferkamp, I. et al. Tapping the nucleotide pool of the host: novel nucleotide carrier proteins of Protochlamydia amoebophila. Mol. Microbiol. 60, 1534–1545 (2006)
Haferkamp, I. 等人。利用宿主的核苷酸库:Protochlamydia amoebophila 的新型核苷酸载体蛋白。微生物。60, 1534–1545 (2006)Miroux, B. & Walker, J. E. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298 (1996)
Miroux, B. & Walker, J. E. 大肠杆菌中蛋白质的过度产生:允许高水平合成一些膜蛋白和球状蛋白的突变宿主。J. Mol. Biol.260, 289–298 (1996 年)Haferkamp, I. & Linka, N. Functional expression and characterisation of membrane transport proteins. Plant Biol. 14, 675–690 (2012)
Haferkamp, I. & Linka, N. 膜转运蛋白的功能表达和表征。植物生物学。14, 675–690 (2012)Ast, M. et al. Diatom plastids depend on nucleotide import from the cytosol. Proc. Natl Acad. Sci. USA 106, 3621–3626 (2009)
Ast, M. 等人。硅藻质体依赖于从胞质溶胶导入的核苷酸。美国国家科学院院刊 106, 3621–3626 (2009)Baba, T. et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008 (2006)
Baba, T. 等人。构建大肠杆菌 K-12 框内单基因敲除突变体:庆应义塾收藏。分子系统生物学。2, 2006.0008 (2006)Lavergne, T., Malyshev, D. A. & Romesberg, F. E. Major groove substituents and polymerase recognition of a class of predominantly hydrophobic unnatural base pairs. Chemistry 18, 1231–1239 (2012)
Lavergne, T., Malyshev, D. A. & Romesberg, F. E. 一类主要疏水性非自然碱基对的主要沟槽取代基和聚合酶识别。化学 18, 1231–1239 (2012)Seo, Y. J., Hwang, G. T., Ordoukhanian, P. & Romesberg, F. E. Optimization of an unnatural base pair toward natural-like replication. J. Am. Chem. Soc. 131, 3246–3252 (2009)
Seo, Y. J., Hwang, GT, Ordoukhanian, P. & Romesberg, F. E. 非自然碱基对向自然类复制的优化。J. Am. Chem. Soc.131, 3246–3252 (2009)Li, L. et al. Natural-like replication of an unnatural base pair for the expansion of the genetic alphabet and biotechnology applications. J. Am. Chem. Soc. 136, 826–829 (2014)
Li, L. 等人。非天然碱基对的类自然复制,用于扩展遗传字母表和生物技术应用。J. Am. Chem. Soc.136, 826–829 (2014 年)Tomizawa, J. & Selzer, G. Initiation of DNA synthesis in Escherichia coli. Annu. Rev. Biochem. 48, 999–1034 (1979)
Tomizawa, J. & Selzer, G. 大肠杆菌中 DNA 合成的启动。Annu. Rev. 生物化学。48, 999–1034 (1979)Allen, J. M. et al. Roles of DNA polymerase I in leading and lagging-strand replication defined by a high-resolution mutation footprint of ColE1 plasmid replication. Nucleic Acids Res. 39, 7020–7033 (2011)
Allen, JM 等人。DNA 聚合酶 I 在 ColE1 质粒复制的高分辨率突变足迹定义的前导链和滞后链复制中的作用。核酸研究39, 7020–7033 (2011)Hashimoto, H. et al. Structure of a Naegleria Tet-like dioxygenase in complex with 5-methylcytosine DNA. Nature 506, 391–395 (2013)
Hashimoto, H. 等人。与 5-甲基胞嘧啶 DNA 复合物中的 Naegleria Tet 样双加氧酶的结构。自然 506, 391–395 (2013)Malyshev, D. A., Seo, Y. J., Ordoukhanian, P. & Romesberg, F. E. PCR with an expanded genetic alphabet. J. Am. Chem. Soc. 131, 14620–14621 (2009)
Malyshev, D. A., Seo, Y. J., Ordoukhanian, P. & Romesberg, F. E. PCR具有扩展的遗传字母表。J. Am. Chem. Soc.131, 14620–14621 (2009)Hirao, I. et al. An unnatural hydrophobic base pair system: site-specific incorporation of nucleotide analogs into DNA and RNA. Nature Methods 3, 729–735 (2006)
Hirao, I. 等人。非自然疏水碱基对系统:核苷酸类似物位点特异性掺入 DNA 和 RNA。自然方法 3, 729–735 (2006)Goodman, M. F. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 71, 17–50 (2002)
Goodman, MF 原核生物和真核生物中易出错的修复 DNA 聚合酶。Annu. Rev. 生物化学。71, 17–50 (2002)Quan, J. & Tian, J. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nature Protocols 6, 242–251 (2011)
Quan, J. & Tian, J. 用于复杂和组合 DNA 文库高通量克隆的环状聚合酶延伸克隆。自然协议 6, 242–251 (2011)Ludwig, J. & Eckstein, F. Rapid and efficient synthesis of nucleoside 5′-0-(1-thiotriphosphates), 5′-triphosphates and 2',3′-cyclophosphorothioates using 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one. J. Org. Chem. 54, 631–635 (1989)
Ludwig, J. & Eckstein, F. 使用 2-氯-4H-1,3,2-苯并二氧杂磷-4-酮快速高效合成核苷 5′-0-(1-硫代三磷酸)、5′-三磷酸和 2',3′-环磷酸盐。J. Org. 化学。54, 631–635 (1989)Alpert, A. & Shukla, A. Precipitation of Large, High-Abundance Proteins from Serum with Organic Solvents in ABRF 2003: Translating Biology using Proteomics and Functional Genomics Poster no. P111-W http://www.abrf.org/Other/ABRFMeetings/ABRF2003/Alpert.pdf (2003)
Alpert, A. & Shukla, A. 在ABRF 2003中用有机溶剂从血清中沉淀出大而高丰度的蛋白质:使用蛋白质组学和功能基因组学翻译生物学海报编号。P111-W http://www.abrf.org/Other/ABRFMeetings/ABRF2003/Alpert.pdf (2003)Kubitschek, H. E. & Friske, J. A. Determination of bacterial cell volume with the Coulter Counter. J. Bacteriol. 168, 1466–1467 (1986)
Kubitschek, H. E. & Friske, J. A. 用库尔特计数器测定细菌细胞体积。J. 细菌。168, 1466–1467 (1986)Yanes, O., Tautenhahn, R., Patti, G. J. & Siuzdak, G. Expanding coverage of the metabolome for global metabolite profiling. Anal. Chem. 83, 2152–2161 (2011)
Yanes, O., Tautenhahn, R., Patti, G. J. & Siuzdak, G. 扩大代谢组的覆盖范围以进行全球代谢物分析。Anal. 化学。83, 2152–2161 (2011)Seidman, C. E., Struhl, K., Sheen, J. & Jessen, T. Introduction of plasmid DNA into cells. Curr. Prot. Mol. Biol Chapter 1, Unit 1.8 (2001)
Seidman, C. E., Struhl, K., Sheen, J. & Jessen, T. 将质粒DNA引入细胞。Curr. Prot. Mol. Biol第 1 章,第 1.8 单元 (2001)Knab, S., Mushak, T. M., Schmitz-Esser, S., Horn, M. & Haferkamp, I. Nucleotide parasitism by Simkania negevensis (Chlamydiae). J. Bacteriol. 193, 225–235 (2011)
Knab, S., Mushak, T. M., Schmitz-Esser, S., Horn, M. & Haferkamp, I. Simkania negevensis(衣原体)的核苷酸寄生。J. 细菌。193, 225–235 (2011 年)Audia, J. P. & Winkler, H. H. Study of the five Rickettsia prowazekii proteins annotated as ATP/ADP translocases (Tlc): Only Tlc1 transports ATP/ADP, while Tlc4 and Tlc5 transport other ribonucleotides. J. Bacteriol. 188, 6261–6268 (2006)
Audia, J. P. & Winkler, H. H. 对五种注释为ATP/ADP转位酶(Tlc)的立克次体蛋白的研究:只有Tlc1运输ATP/ADP,而Tlc4和Tlc5运输其他核糖核苷酸。J. 细菌。188, 6261–6268 (2006 年)Hofer, A., Ekanem, J. T. & Thelander, L. Allosteric regulation of Trypanosoma brucei ribonucleotide reductase studied in vitro and in vivo. J. Biol. Chem. 273, 34098–34104 (1998)
Hofer, A., Ekanem, J. T. & Thelander, L. 在体外和体内研究了布鲁氏锥虫核糖核苷酸还原酶的变构调节。J. Biol. 化学。273, 34098–34104 (1998)Reijenga, J. C., Wes, J. H. & van Dongen, C. A. M. Comparison of methanol and perchloric acid extraction procedures for analysis of nucleotides by isotachophoresis. J. Chromatogr. B Biomed. Sci. Appl. 374, 162–169 (1986)
Reijenga, J. C., Wes, J. H. & van Dongen, C. A. M. 通过等速泳分析核苷酸的甲醇和高氯酸萃取程序的比较。J. 色谱仪。B 生物医学。科学应用。374, 162–169 (1986 年)
Acknowledgements 确认
We thank I. Haferkamp and J. Audia for kindly providing the NTT plasmids and helpful discussions, and P. Ordoukhanian for providing access to the Center for Protein and Nucleic Acid Research at TSRI. This work was supported by the US National Institutes of Health (NIH) (GM 060005).
我们感谢 I. Haferkamp 和 J. Audia 慷慨地提供 NTT 质粒和有益的讨论,并感谢 P. Ordoukhanian 提供对 TSRI 蛋白质和核酸研究中心的访问。这项工作得到了美国国立卫生研究院 (NIH) (GM 060005) 的支持。
Author information 作者信息
Authors and Affiliations
Contributions
D.A.M., K.D., T.C. and F.E.R. designed the experiments. D.A.M., K.D. and T.L. performed the experiments. N.D., J.M.F. and I.R.C.J. performed LC-MS/MS analysis. D.A.M., K.D. and F.E.R. analysed data and D.A.M. and F.E.R. wrote the manuscript with assistance from the other authors.
Corresponding author
Ethics declarations 道德宣言
Competing interests 利益争夺
F.E.R. and D.A.M. have filed a patent application based on the use of NTTs for biotechnological applications. F.E.R. D.A.M., T.L. and K.D. have shares in Synthorx Inc., a company that has commercial interests in the UBP. D.A.M. and K.D. are currently employed by Synthorx Inc. The other authors declare no competing financial interests.
F.E.R. 和 D.A.M. 已经提交了一项基于 NTT 用于生物技术应用的专利申请。F.E.R. D.A.M.、T.L. 和 K.D. 持有 Synthorx Inc. 的股份,该公司在 UBP 中拥有商业利益。D.A.M. 和 K.D. 目前受雇于 Synthorx Inc.。其他作者声明没有相互竞争的经济利益。
Extended data figures and tables
扩展数据图表和表格
Extended Data Figure 1 Natural triphosphate uptake by NTTs.
扩展数据 图 1 NTT 对三磷酸盐的天然摄取。
a, Survey of reported substrate specificity (KM, μM) of the NTTs assayed in this study. b, PtNTT2 is significantly more active in the uptake of [α-32P]-dATP compared to other nucleotide transporters. Raw (left) and processed (right) data are shown. Relative radioactivity corresponds to the total number of counts produced by each sample. Interestingly, both PamNTT2 and PamNTT5 exhibit a measurable uptake of dATP although this activity was not reported before. This can possibly be explained by the fact that substrate specificity was only characterized using competition experiments, and assay sensitivity might not have been adequate to detect this activity15. References 35, 36 are cited in this figure.
a,本研究中测定的 NTT 的底物特异性 (KM, μM) 的调查。b,与其他核苷酸转运蛋白相比,PtNTT2 在 [α-32P]-dATP 的摄取中显着更活跃。显示原始 (左) 和已处理 (右) 数据。相对放射性对应于每个样品产生的计数总数。有趣的是,PamNTT2 和 PamNTT5 都表现出可测量的 dATP 摄取,尽管这种活性以前没有报道。这可能可以通过以下事实来解释:底物特异性仅使用竞争实验进行表征,并且检测灵敏度可能不足以检测这种活性15。本图引用了参考文献 35、36。
Extended Data Figure 2 Degradation of unnatural triphosphates in growth media.
扩展数据图 2 生长培养基中非天然三磷酸盐的降解。
Unnatural triphosphates (3P) of dNaM and d5SICS are degraded to diphosphates (2P), monophosphates (1P) and nucleosides (0P) in the growing bacterial culture. Potassium phosphate (KPi) significantly slows down the dephosphorylation of both unnatural triphosphates. a, Representative HPLC traces (for the region between ∼20 and 24 min). dNaM and d5SICS nucleosides are eluted at approximately 40 min and not shown. b, Composition profiles.
在生长的细菌培养物中,dNaM 和 d5SICS 的非天然三磷酸盐 (3P) 被降解为二磷酸盐 (2P)、单磷酸盐 (1P) 和核苷 (0P)。磷酸钾 (KPi) 显着减慢两种非天然三磷酸盐的去磷酸化。a,代表性的 HPLC 迹线(对于 ∼20 到 24 分钟之间的区域)。dNaM 和 d5SICS 核苷在大约 40 分钟时洗脱,未显示。b,合成配置文件。
Extended Data Figure 3 Effect of potassium phosphate on dATP uptake and stability in growth media.
扩展数据图 3 磷酸钾对生长培养基中 dATP 摄取和稳定性的影响。
a, KPi inhibits the uptake of [α-32P]-dATP at concentrations above 100 mM. Raw (left) and processed (right) data are shown. The NTT from Rickettsia prowazekii (RpNTT2) does not mediate the uptake of any of the dNTPs and was used as a negative control: its background signal was subtracted from those of PtNTT2 (black bars) and TpNTT2 (white bars). Relative radioactivity corresponds to the total number of counts produced by each sample. b, KPi (50 mM) significantly stabilizes [α-32P]-dATP in the media. Triphosphate stability in the media is not significantly affected by the nature of the NTT expressed. 3P, 2P and 1P correspond to triphosphate, diphosphate and monophosphate states, respectively. Error bars represent s.d. of the mean, n = 3.
a,KPi 抑制浓度高于 100 mM 的 [α-32P]-dATP 的摄取。显示原始 (左) 和已处理 (右) 数据。来自普罗瓦泽基立克次体 (RpNTT2) 的 NTT 不介导任何 dNTP 的摄取,并用作阴性对照:从 PtNTT2 (黑条) 和 TpNTT2 (白条) 的背景信号中减去其背景信号。相对放射性对应于每个样品产生的计数总数。b,KPi (50 mM) 显着稳定培养基中的 [α-32P]-dATP。培养基中的三磷酸盐稳定性不受表达的 NTT 性质的显著影响。3P、2P 和 1P 分别对应于三磷酸盐、二磷酸盐和单磷酸盐状态。误差线表示平均值的 s.d.,n = 3。
Extended Data Figure 4 dATP uptake and growth of cells expressing PtNTT2 as a function of inducer (IPTG) concentration.
扩展数据图 4 表达 PtNTT2 的细胞的 dATP 摄取和生长与诱导剂 (IPTG) 浓度的函数关系。
Growth curves and [α-32P]-dATP uptake by bacterial cells transformed with pCDF-1b-PtNTT2 (pACS) plasmid as a function of IPTG concentration. a, Total uptake of radioactive substrate (left) and total intracellular triphosphate content (right) are shown at two different time points. Relative radioactivity corresponds to the total number of counts produced by each sample. b, A stationary phase culture of C41(DE3) pACS cells was diluted 100-fold into fresh 2 × YT media containing 50 mM KPi, streptomycin, and IPTG at the indicated concentrations and were grown at 37°C. Error bars represent s.d. of the mean, n = 3.
用 pCDF-1b-Pt NTT2 (pACS) 质粒转化的细菌细胞的生长曲线和 [α-32P]-dATP 摄取与 IPTG 浓度的关系。a,放射性底物的总摄取量(左)和细胞内总三磷酸盐含量(右)显示在两个不同的时间点。相对放射性对应于每个样品产生的计数总数。b,将 C41 (DE3) pACS 细胞的固定相培养物稀释 100 倍,放入含有指定浓度的 50 mM KPi、链霉素和 IPTG 的新鲜 2 × YT 培养基中,并在 37°C 下生长。 误差线表示平均值的 s.d.,n = 3。
Extended Data Figure 5 Stability and uptake of dATP in the presence of 50 mM KPi and 1 mM IPTG.
扩展数据 图 5 在 50 mM KPi 和 1 mM IPTG 存在下 dATP 的稳定性和摄取。
Composition of [α-32P]-dATP in the media (left) and cytoplasmic fraction (right) as a function of time. TLC images and their quantifications are shown at the bottom and the top of each of the panels, respectively. 3P, 2P and 1P correspond to nucleoside triphosphate, diphosphate and monophosphate, respectively. M refers to a mixture of all three compounds that was used as a TLC standard. The position labelled ‘Start’ corresponds to the position of sample spotting on the TLC plate.
培养基中 [α-32P]-dATP 的组成(左)和细胞质组分(右)随时间的变化。TLC 图像及其定量分别显示在每个面板的底部和顶部。3P、2P 和 1P 分别对应于核苷三磷酸、二磷酸和单磷酸。M 是指用作 TLC 标准品的所有三种化合物的混合物。标有“开始”的位置对应于 TLC 板上样品点样的位置。
Extended Data Figure 6 Calibration of the streptavidin shift (SAS).
扩展数据图 6 链霉亲和素转移 (SAS) 的校准。
a, The SAS is plotted as a function of the fraction of template containing the UBP. Error bars represent s.d. of the mean, n = 3. b, Representative data. SA, streptavidin.
a,SAS 绘制为包含 UBP 的模板分数的函数。误差线表示平均值的 s.d.,n = 3。乙,代表性数据。SA,链霉亲和素。
Extended Data Figure 7 Decomposition of unnatural triphosphates, pINF quantification, and retention of the UBP with extended cell growth.
扩展数据 图 7 非天然三磷酸盐的分解、pINF 定量和随着细胞生长延长而保留 UBP。
a, Dephosphorylation of the unnatural nucleoside triphosphate. 3P, 2P, 1P and 0P correspond to triphosphate, diphosphate, monophosphate and nucleoside states, respectively. The composition at the end of the 1 h recovery is shown at the right. b, Restriction analysis of pINF and pACS plasmids purified from E. coli, linearized with NdeI restriction endonuclease and separated on a 1% agarose gel (assembled from independent gel images). Molar ratios of pINF/pACS plasmids are shown at the top of each lane. For each time point, triplicate data are shown in three lanes with the untransformed control shown in the fourth, rightmost lane (see Methods). c, Number of pINF doublings as a function of time. The decrease starting at approximately 50 h is due to the loss of the pINF plasmid that also results in increased error. See the section on pINF replication in E. coli in the Methods for details. d, UBP retention (%) as a function of growth as determined by gel shift (data shown in Fig. 3) and Sanger sequencing (sequencing traces are available as Supplementary Data). In a, c and d, error shown is the s.d. of mean, n = 3.
a,非天然核苷三磷酸的去磷酸化。3P、2P、1P 和 0P 分别对应于三磷酸盐、二磷酸盐、单磷酸盐和核苷状态。1 小时恢复结束时的成分如右图所示。b,对从大肠杆菌中纯化的 pINF 和 pACS 质粒进行限制性分析,用 NdeI 限制性核酸内切酶线性化,并在 1% 琼脂糖凝胶上分离(由独立的凝胶图像组装而成)。pINF/pACS 质粒的摩尔比显示在每个泳道的顶部。对于每个时间点,一式三份数据显示在三个通道中,未转换的控件显示在最右侧的第四个通道中(参见方法)。c, pINF 倍增次数随时间的变化。从大约 50 小时开始的减少是由于 pINF 质粒的丢失,这也导致误差增加。有关详细信息,请参阅方法中关于大肠杆菌中 pINF 复制的部分。d,UBP 保留率 (%) 与生长的关系,由凝胶位移(数据如图 3 所示)和 Sanger 测序(测序轨迹作为补充数据提供)确定。在 a、c 和 d 中,显示的误差是平均值 n = 3 的 s.d.。
扩展数据表 1 大肠杆菌培养物的 OD600 和质粒(pINF 或对照 pUC19)的相对拷贝数,由生长 19 小时后与 pACS 的摩尔比确定
扩展数据表 2 使用含有 d5SICS 和 dNaM 的合成寡核苷酸通过 LC-MS/MS 进行相对定量
扩展数据表 3 最成功的提取方法总结
Supplementary information
Supplementary Information
The file contains the sequences of oligonucleotides used in this study, an example calculation of plasmid amplification, and the sequence of the pACS plasmid. (PDF 243 kb)
Supplementary Data
This file contains raw sequencing traces for PCR fragments generated from pINF plasmid propagated in E. coli at different time points (n=3). The position of the unnatural nucleotide is indicated with a red arrow. (XLSX 363 kb)
Rights and permissions
About this article

Cite this article
Malyshev, D., Dhami, K., Lavergne, T. et al. A semi-synthetic organism with an expanded genetic alphabet. Nature 509, 385–388 (2014). https://doi.org/10.1038/nature13314
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature13314
This article is cited by
-
SnIP-type atomic-scale inorganic double-helix semiconductors: Synthesis, properties, and applications
Nano Research (2024)
-
Quintuply orthogonal pyrrolysyl-tRNA synthetase/tRNAPyl pairs
Nature Chemistry (2023)
-
How scientists are hacking the genetic code to give proteins new powers
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.