
摘要
The Janus kinase (JAK) signal transducer and activator of transcription (JAK-STAT) pathway is an evolutionarily conserved mechanism of transmembrane signal transduction that enables cells to communicate with the exterior environment. Various cytokines, interferons, growth factors, and other specific molecules activate JAK-STAT signaling to drive a series of physiological and pathological processes, including proliferation, metabolism, immune response, inflammation, and malignancy. Dysregulated JAK-STAT signaling and related genetic mutations are strongly associated with immune activation and cancer progression. Insights into the structures and functions of the JAK-STAT pathway have led to the development and approval of diverse drugs for the clinical treatment of diseases. Currently, drugs have been developed to mainly target the JAK-STAT pathway and are commonly divided into three subtypes: cytokine or receptor antibodies, JAK inhibitors, and STAT inhibitors. And novel agents also continue to be developed and tested in preclinical and clinical studies. The effectiveness and safety of each kind of drug also warrant further scientific trials before put into being clinical applications. Here, we review the current understanding of the fundamental composition and function of the JAK-STAT signaling pathway. We also discuss advancements in the understanding of JAK-STAT–related pathogenic mechanisms; targeted JAK-STAT therapies for various diseases, especially immune disorders, and cancers; newly developed JAK inhibitors; and current challenges and directions in the field.
Janus 激酶(JAK)信号转导和转录激活因子(JAK-STAT)通路是一种进化保守的跨膜信号转导机制,可使细胞与外界环境进行交流。各种细胞因子、干扰素、生长因子和其他特定分子会激活 JAK-STAT 信号,从而驱动一系列生理和病理过程,包括增殖、新陈代谢、免疫反应、炎症和恶性肿瘤。失调的 JAK-STAT 信号和相关基因突变与免疫激活和癌症进展密切相关。对 JAK-STAT 通路的结构和功能的深入研究促使人们开发并批准了多种用于临床疾病治疗的药物。目前,主要针对 JAK-STAT 通路开发的药物通常分为三个亚型:细胞因子或受体抗体、JAK 抑制剂和 STAT 抑制剂。新型药物也在不断开发,并在临床前和临床研究中进行测试。在投入临床应用之前,每种药物的有效性和安全性还需要进一步的科学试验。在此,我们回顾了目前对 JAK-STAT 信号通路的基本组成和功能的理解。我们还将讨论对 JAK-STAT 相关致病机制认识的进展;针对各种疾病(尤其是免疫性疾病和癌症)的 JAK-STAT 靶向疗法;新开发的 JAK 抑制剂;以及该领域目前面临的挑战和发展方向。
Similar content being viewed by others
他人正在浏览的类似内容

Introduction 导言
The Janus kinase (JAK) signal transducer and activator of transcription (JAK-STAT) pathway is an evolutionarily conserved signaling pathway that functions in several crucial physiological processes, including hematopoiesis, differentiation, metabolism, and immune modulation.1,2,3,4 Structurally, the JAK-STAT pathway involves transmembrane receptors, receptor-associated cytosolic tyrosine kinases (i.e. JAKs), and signal transducers and activators of transcription (i.e., STATs).5 The JAK protein family contains four members: JAK1, JAK2, JAK3, and TYK2.6,7,8,9 The STAT family consists of seven proteins: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6.10,11,12 The JAK-STAT signaling pathway was first discovered in investigations of interferon-related transcriptional activation. Subsequently, a general outline of the components and pathogenesis of the JAK-STAT signaling pathway was gradually completed over a period of about 20 years.13,14,15 More than 50 types of cytokines, including interferons (IFNs), interleukins (ILs), and growth factors, have been shown to play roles in JAK-STAT signaling to fulfill regulatory functions in cell differentiation, metabolism, survival, homeostasis, and immune response. Once receptors bind to an extracellular ligand, JAKs initiate tyrosine phosphorylation of the receptors and recruit corresponding STATs.16,17,18 The phosphorylated STATs then dimerize and enter the nucleus to regulate specific gene transcription. This process enables the rapid transmission of external signals to the nucleus to regulate biological and pathological processes. Genome-wide association studies for disease exploration have identified more than 200 somatic mutations and single-nucleotide polymorphisms of JAK-STAT pathway genes that are functionally correlated with human diseases, including rheumatoid arthritis (RA), hematological malignancies, and atopic dermatitis (AD).19,20,21 Abnormal activation of JAK-STAT signaling has been identified in diverse immune-mediated conditions and cancers, including melanomas, glioblastomas, and head, neck, lung, pancreatic, breast, rectal, and prostate cancers.22,23,24,25,26,27 Based on the ‘double-edged sword’ function of the JAK-STAT pathway in disease pathogenesis, numerous agents targeting the JAK-STAT pathway have been developed and tested in preclinical and clinical trials.28,29,30,31 As a result, first-generation JAK inhibitors, such as ruxolitinib, tofacitinib, and baricitinib, have been approved for clinical use.32,33,34 Furthermore, a new generation of selective JAK inhibitors has emerged as a promising option for drug development and has shown success in preclinical trials. Despite this progress, some studies have reported adverse effects of JAK inhibitors, including infection, hematologic events, Wernicke encephalopathy, and even cancer, emphasizing the need for follow-up studies and in-depth investigations on drug thresholds, the mechanisms of adverse events, and drug resistance.35,36,37
Janus 激酶(JAK)信号转导和转录激活因子(JAK-STAT)通路是一条进化保守的信号通路,在造血、分化、新陈代谢和免疫调节等多个关键生理过程中发挥作用。从结构上看,JAK-STAT 通路涉及跨膜受体、与受体相关的细胞膜酪氨酸激酶(即 JAKs)以及信号转导和转录激活因子(即 STATs)5:5 JAK 蛋白家族包括四个成员:JAK1、JAK2、JAK3 和 TYK2。6,7,8,9STAT 家族包括七个蛋白:STAT1、STAT2、STAT3、STAT4、STAT5A、STAT5B 和 STAT6。13,14,15包括干扰素(IFNs)、白细胞介素(ILs)和生长因子在内的 50 多种细胞因子已被证明在 JAK-STAT 信号传导中发挥作用,在细胞分化、新陈代谢、存活、稳态和免疫反应中发挥调节功能。 一旦受体与细胞外配体结合,JAKs 就会启动受体的酪氨酸磷酸化,并招募相应的 STATs。这一过程可使外部信号快速传递到细胞核,从而调节生物和病理过程。为探索疾病而进行的全基因组关联研究已发现 200 多个 JAK-STAT 通路基因的体细胞突变和单核苷酸多态性与人类疾病(包括类风湿性关节炎 (RA)、血液系统恶性肿瘤和特应性皮炎 (AD))存在功能相关性。19,20,21在多种免疫介导的疾病和癌症中,包括黑色素瘤、胶质母细胞瘤、头颈癌、肺癌、胰腺癌、乳腺癌、直肠癌和前列腺癌,都发现了 JAK-STAT 信号的异常激活。基于 JAK-STAT 通路在疾病发病机制中的 "双刃剑 "功能,许多针对 JAK-STAT 通路的药物已被开发出来,并在临床前和临床试验中进行了测试。32,33,34此外,新一代的选择性 JAK 抑制剂已成为药物开发的一个有前途的选择,并在临床前试验中取得了成功。尽管取得了这一进展,但仍有一些研究报告了 JAK 抑制剂的不良反应,包括感染、血液学事件、Wernicke 脑病,甚至癌症,这强调了对药物阈值、不良反应机制和耐药性进行后续研究和深入调查的必要性。35,36,37
在这篇综述中,我们在以往研究的基础上,扩展并更新了有关 JAK-STAT 信号转导的组成、经典激活和负调控的综合研究。我们特别强调了 JAK-STAT 信号的免疫调节作用以及 JAK-STAT 信号通路与某些疾病之间的遗传关联。我们还详细讨论了 JAK 抑制剂在治疗免疫相关疾病和恶性疾病中的应用,并总结了其在一系列临床和临床前试验中的有效性和安全性。
JAK、STAT 和 JAK-STAT 通路
JAK-STAT 通路是一种进化保守的信号通路,它由细胞因子刺激激活,使细胞外信号穿过细胞膜传递到细胞核,引起 DNA 转录的变化。
JAK 家族
非受体酪氨酸激酶 JAK 家族由四种蛋白组成:48、49、50、51、52、53JAK3 主要在造血细胞中高水平表达,而其他成员则在许多组织中广泛表达。59FERM 和 SH2 结构域主要负责 JAK 与受体的结合。伪激酶结构域调节激酶结构域的活性,激酶结构域对受体酪氨酸的磷酸化至关重要,而受体酪氨酸的磷酸化又会导致下游分子的进一步磷酸化。60这四个结构域又可分为七个部分,称为 JH1-7。63,64,65,66,67JH1编码一种激酶,可将底物激酶结构域的一个重要组成部分磷酸化。68,69,70JH2(也称为无激酶活性的伪激酶结构域)的主要功能是增强JH1的激酶功能。JH3和JH4维持激酶结构的稳定性。71,72,73,74,75,76JH5、JH6和JH7负责将JAK连接到相应的受体上。细胞因子(如干扰素、白介素和生长因子)及其受体是 JAK 的主要激活剂。77受体-配体复合物激活与受体结合的 JAK,JAK 催化受体酪氨酸磷酸化。78,79,80JAK1、JAK3 和 TYK2 负责免疫系统的发育和免疫调节,而 JAK2 则主要参与造血。81,82,83,84,85,86,87,88,89,90
STAT 家庭
STAT 蛋白是 JAK 下游的信号分子。STAT 家族成员包括 STAT1、STAT2、STAT3、STAT4、STAT5A、STAT5B 和 STAT6。91,92,93,94STATs 由一个 N 端结构域和线圈、一个螺旋结构域、一个 DNA 结合结构域、一个连接结构域、一个 SH2 结构域和一个转录激活结构域组成。101、102、103、104DNA 结合结构域可使 STAT 作为转录因子与 DNA 结合。受体酪氨酸被磷酸化后,细胞质中的 STATs 被招募到活化的受体上,STAT 酪氨酸被磷酸化,从而形成 STAT 二聚体。107,108,109STAT 二聚体随后作为转录因子复合物的成分进入细胞核,促进特定靶基因的转录。 在 STAT 家族中,STAT3 被认为在从质膜到细胞核的信号传递过程中发挥着核心作用,因此是一个很有希望的药物开发靶点。112,113,114
JAK-STAT 信号的负调控
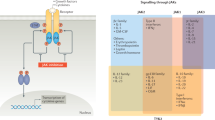
许多调节因子可抑制 JAK-STAT 信号通路的激活。JAK-STAT 调节因子主要有三种:细胞因子信号转导抑制因子(SOCS)、活化 STAT 蛋白抑制因子(PIAS)和蛋白酪氨酸磷酸酶。SOCS 家族是减弱 JAK-STAT 通路的主要信号分子,包括 CIS、SOCS1、SOCS2、SOCS3、SOCS4、SOCS5、SOCS6 和 SOCS7。SOCS 可由 IL-2、IL-3 和 IFN-γ 等细胞因子诱导。活化的 STAT 进入细胞核会增强 SOCS 的转录,SOCS 通过阻断 STAT 受体结合、通过 N 端激酶抑制结构使 JAK 失活或结合并泛素化 JAK 或 STAT 以进行蛋白酶体降解,从而对 JAK-STAT 信号转导产生负向调节作用123,124。125PIAS 家族包括 PIAS1、PIAS3、PIASx 和 PIASy。126,127,128PIASs 可与 STAT 相互作用,阻止 STAT 二聚化或阻止 STAT 二聚体与 DNA 结合。作为磷酸酶,蛋白酪氨酸磷酸酶可与受体相互作用,使 JAK 去磷酸化。 蛋白酪氨酸磷酸酶还能直接使 STAT 二聚体去磷酸化,从而抑制 JAK-STAT 信号传导(图1)。129,130

JAK-STAT信号通路的典型激活和负调控。JAK-STAT信号通路的典型激活:细胞因子与其相应的受体结合,受体发生构象变化,并招募相关的JAKs。JAK 发生磷酸化,从而导致受体发生酪氨酸磷酸化,为 STAT 创造对接位点。STAT 磷酸化后与受体分离,以同二聚体或异二聚体形式进入细胞核,与特定 DNA 位点结合,调节细胞因子相关基因的转录。JAK-STAT 信号通路的负调控:CIS/SOCS(细胞因子信号抑制剂)、PIAS(活化 STAT 蛋白抑制剂)和 PTP(蛋白酪氨酸磷酸酶)家族参与了 JAK-STAT 信号通路的负调控。CIS/SOCS家族通过直接结合JAKs或JAK受体抑制JAK激酶活性、结合受体酪氨酸激酶位点拦截STAT-受体结合或形成酶复合物降解JAKs或STAT等方式对JAK-STAT通路进行负调控。PIAS 家族主要与 STAT 二聚体相互作用,抑制 STAT 与 DNA 的结合,从而阻断 JAK-STAT 信号转导。PTP 家族主要通过使活化的受体、JAKs 和 STATs 去磷酸化来负向调节 JAK/STAT 通路。实线表示激活过程。虚线表示负调控
JAK-STAT通路、免疫调节和血统可塑性
细胞因子对体液免疫和细胞免疫反应至关重要。44,131,132与自身免疫疾病相关的多种细胞因子,包括 IFNs、ILs 和集落刺激因子,主要通过 I 型和 II 型细胞因子受体的组合发挥多向效应。1,136,137大量研究表明,细胞因子诱导的 JAK-STAT 通路激活在免疫细胞的分化和发育以及免疫系统的稳态中发挥着重要作用(图2)。138,139,140在不同细胞因子的刺激下,JAK-STAT 信号通路在免疫调节事件中发挥复杂的功能,这些功能不仅包括主要由 IFNs 和 STAT1、STAT2 信号引发的癌细胞识别,还包括主要由 IL-6-STAT3 信号引发的免疫逃逸。研究广泛表明,JAK-STAT 信号通过诱导自然杀伤(NK)细胞的活化、细胞毒性和功能,在细胞因子(如 IL-2、IL-15 和 IFNs)的作用下驱动抗肿瘤免疫监视。141 ,142 ,143,144另一方面,JAK-STAT 通路也与许多自身免疫性疾病(如 RA、炎症性肠病和 AD)的发病机制有关。148在脊柱关节炎(Spondyloarthritis,SpA)动物模型中,TYK2 被证明是 IL-22 诱导的 STAT3 磷酸化的重要介质,可增强 3 型免疫并加速 SpA 的发展149。此外,多项全基因组关联分析清楚地表明,JAK-STAT 通路的多态性和突变与自身免疫性疾病和免疫介导的癌症有关(图3)。Fabre, A. 等人报告了 STAT3 功能增益突变与早发多自身免疫之间的联系。153也有报道称,突变的 STAT3 通过改变 T 细胞表型并赋予 CD8+ T 细胞细胞毒性特性,参与了免疫介导的再生障碍性贫血的发病机制。154此外,还发现 JAK 突变可阻断 IFN-γ 信号传递,导致免疫逃避和对抗病毒-1/PD-L1 免疫疗法不敏感。最近的临床试验表明,以 JAK 为靶点并阻止其磷酸化可抑制细胞因子引起的异常免疫和炎症反应,这为将 JAK 抑制剂用作治疗自身免疫性疾病和癌症的疗法提供了合理而确凿的证据。
表 1 与 JAK 和 STAT 相关的细胞因子和疾病

JAK-STAT 信号通路与免疫。大量细胞因子与 JAK-STAT 通路之间的相互作用影响着免疫细胞的分化和发育,并产生免疫调节作用。IFN 和 IL-12 对 Th1 细胞的分化至关重要,它们分别通过 STAT1 和 STAT4 驱动 T-bet 基因的表达。IL-4 通过 STAT6 上调 GATA3 基因,激活 Th2 细胞分化。IL-6和TGF通过STAT3触发RORt基因表达,在Th17细胞分化中发挥重要作用。IL-6和IL-12通过STAT3影响T滤泡辅助细胞的分化,从而增加Bcl-6基因的转录。IL-2通过STAT5A/B与Foxp3基因的直接结合诱导Treg细胞分化

人类疾病中的 JAK-STAT 通路突变。JAK-STAT 通路基因的基因突变和多态性广泛涉及人类疾病的发病机制。最常见的致病基因突变位于 JAK 和 STAT 的 SH2 结构域内。
JAK-STAT 通路对疾病进展的影响十分复杂。细胞的可塑性使细胞能够根据环境变化采用新的表型。156,157,158癌细胞会放大这种可塑性,导致肿瘤异质性、转移和耐药性。携带表皮生长因子受体(EGFR)突变的肺癌从腺癌到侵袭性神经内分泌癌的组织学转变是癌症细胞系可塑性的一个显著例子。这方面最突出的例子之一是,活化的JAK-STAT信号在前列腺癌从腺癌向神经内分泌癌的谱系转变过程中做出了贡献。在肝癌中,JAK-STAT3 参与了 RAS 诱导的、与恶性肿瘤相关的肝细胞向肝内胆管癌细胞的转分化167。此外,已知 WNT5A 激活 IL-6-JAK-STAT 通路可促进瘢痕上皮-间质转化(EMT)。160更全面地了解 JAK-STAT 信号通路对细胞系可塑性的影响,将为开发治疗恶性疾病的新疗法提供合理的分子基础。168
JAK-STAT 通路与疾病
越来越多的证据表明,JAK-STAT 激活可在疾病中发挥双重作用。JAK-STAT通路的过度激活与许多疾病的不良预后有关,包括黑色素瘤、胶质母细胞瘤、头颈部癌症、肺癌、胰腺癌、乳腺癌、直肠癌和前列腺癌。在接下来的章节中,我们将重点介绍 JAK-STAT 通路在一些常见疾病中的关键作用,包括类风湿性关节炎、骨髓增生性肿瘤、肾脏疾病以及前列腺癌、乳腺癌和肺癌。
类风湿性关节炎
170,171,172RA 发病机制的一个核心特征是滑膜细胞产生 TNF、IL-1 和 IL-6 等炎性细胞因子。176,177,178,179成纤维细胞是 RA 病变滑膜中炎症的主要驱动因素,已被证实可在 STAT4 激活的情况下产生 IL-6,从而引发持续的关节破坏180。Mori T. 等人指出,IL-6-STAT3 细胞因子环路被 RA 中高表达的炎性细胞因子激活,导致慢性持续性炎症和关节破坏。181活动性 RA 患者的成纤维细胞样滑膜细胞显示 STAT1 表达和活性升高。 JAK3、STAT4 和STAT6也在 RA 患者的 CD1a+树突状细胞中高表达,可能有助于在滑膜水平识别 RA。190另一项研究表明,miR-17 可抑制 IL-6 和 IL-1β 等促炎细胞因子的分泌,并与 STAT3 和 JAK1 结合,在 RA 发病过程中发挥抗炎和抗蚀作用。Wang, J. 等人报道了长基因间非蛋白编码长链 RNA p53 诱导转录本(LncRNA LINC-PINT)通过上调 SOCS1 水平抑制 TNF-α 诱导的 RA 滑膜成纤维细胞。191

JAK-STAT 通路在类风湿关节炎发病机制中的作用以及最近批准的 JAK 抑制剂。多种细胞因子诱导的异常 JAK-STAT 信号传导被认为是 RA 的基本发病机制。多种细胞因子(包括 IL-1、IL-17、IL-12、IL-23、IL-6、TNF-α 和 IFN-γ)与 JAK-STAT 通路相互作用,介导滑膜的炎症反应并导致关节破坏。目前有多种 JAK 抑制剂被批准用于治疗 RA,包括托法替尼、巴利替尼、非格替尼、乌达替尼、培菲替尼和去诺替尼。虚线表示负调控
JAK抑制剂是最近推出的一种新型改善病情抗风湿药物(DMARDs),可减轻滑膜炎和全身炎症,改善RA的功能(表2)。欧洲风湿病学会(EULAR)于 2019 年将 JAK 抑制剂的应用等同于在传统合成 DMARDs(csDMARDs)无效的情况下应用生物 DMARDs(bDMARDs)。192,193,194一项关于常用二线 RA 药物疗效的大型观察性研究证实,JAK 抑制剂比甲氨蝶呤更有效。Chen, C.等人最近提出,JAK3抑制剂Z583与JAK3的半胱氨酸残基909(Cys909)不可逆结合,阻止了JAK-STAT信号的激活,对RA的进展产生了强大的抑制作用。鉴于 JAK-STAT 信号在 RA 中的表达和影响,JAK 抑制剂被认为是治疗 RA 的适当药物,尽管还需要进一步的大规模研究来确定针对 JAK-STAT 信号的药物在 RA 中更具体的临床应用。
表 2 用于治疗 RA 的四种获准 JAKis
骨髓增生性肿瘤
206,207,208,209MPNs 是一种克隆性造血疾病,其特点是髓系细胞过度增殖,导致外周血细胞数量和形态异常,并具有发展为急性髓系白血病的高风险。高分辨率全基因组基因分型发现,在各种类型的多发性骨髓瘤中,获得性体细胞突变与 JAK-STAT 活性增加有关。许多研究报告称,JAK2 突变在多发性骨髓瘤中很常见,并对多种生物过程的调控产生破坏性影响。Rampal R 等人采用综合基因组分析发现,JAK-STAT 靶基因的独特上调有助于区分不同亚型的 MPN,而 JAK2(JAK2V617F)氨基酸617位的缬氨酸-苯丙氨酸置换程度会影响疾病的严重程度223。61,224,225,226Pf4-Cre 转基因小鼠的实验表明,JAK2V617F突变的巨核细胞中激活的 JAK-STAT 信号通过产生促炎细胞因子和趋化因子,促进了 MPN 中骨髓增殖的诱导和维持。基于JAK2V617F与多发性骨髓瘤之间的关联,世界卫生组织分类和国际共识分类都提出了 "JAK2 基因突变高发多发性骨髓瘤 "的新定义,主要包括真性多血细胞增多症、原发性血小板增多症、原发性骨髓纤维化和无法分类的多发性骨髓瘤。基因组分析表明,90% 以上的真性红细胞增多症病例以及约 50% 的原发性血小板增多症和原发性骨髓纤维化病例经常出现JAK2V617F突变。
钙网蛋白(CALR)是一种定位于内质网并参与蛋白质折叠的伴侣蛋白,是继 JAK2 之后 MPN 中第二种最常见的突变蛋白。CALR突变与其他MPN驱动突变(包括JAK2V617F和血小板生成素受体(MPL)突变MPLW515L)相互排斥,这一点已得到公认229,230,231。全外显子组测序在约 70% 至 80% 的 JAK2V617F阴性 ET 和原发性骨髓纤维化中发现了 CALR 突变。232,233体内研究表明,仅 CALR 突变就足以启动 MPN 表型并增加 JAK-STAT 信号活性。细胞模型显示,CALR 突变体能特异性激活 MPL,驱动 MPN 的发病机制。Elf, S. 等人进一步证明,MPN 中突变 CALR 驱动的恶性转化需要与 MPL 相互作用。
血小板生成素及其受体 MPL 是巨核细胞生长和分化的主要调节因子。236,237,238 MPLW515L的致病突变会诱导 JAK-STAT 信号的异常激活,并参与 MPN 的形成。Lnk 是血小板生成素的负调控因子,可逆转携带MPLW515L的细胞中的生长刺激信号,导致恶性细胞死亡。此外,一项针对 1182 名 MPN 患者的研究以及对不同病程的骨髓衍生 DNA 的研究一致表明,MPLW515L和JAK2V617F在 MPN 中同时出现,这表明这些驱动突变的功能在 MPN 发病过程中可能是相对互补的240,241。
特应性皮炎
特应性皮炎(AD)是最常见的慢性免疫介导型皮肤病之一。242,243,244现有的特应性皮炎疗法能缓解症状,但不能延缓疾病的发展,而且所有这些疗法在临床应用中都有局限性。245,246特应性皮炎的发病机制有多种因素,主要是由 T 辅助细胞 2(Th2)介导的炎症以及疾病发展后期 Th1、Th17 和 Th22 细胞介导的炎症逐渐上调所致247。包括 IL-4、IL-13、IL-31 和 TSLP 在内的多种介质与特定跨膜受体结合并刺激 JAK-STAT 通路,从而启动细胞内信号传导并在 AD 中发挥促炎作用。248,249对 AD 发病机制的深入研究表明,JAK-STAT 通路参与了 AD 进展过程中的 Th2 免疫极化、嗜酸性粒细胞活化和皮肤屏障破坏。
随着人们对 JAK-STAT 通路在 AD 中的作用有了更深入的了解,JAK 抑制剂已成为治疗 AD 的一种新方法。251,252,253巴比替尼是一种口服的 JAK1 和 JAK2 选择性抑制剂,已被批准用于治疗中度至重度 AD。两项三期试验(BREEZE-AD1 和 BREEZE-AD2)的结果表明,巴利昔替尼可明显缓解中重度 AD 患者难以忍受的瘙痒和皮肤损伤。在实验模型和 3 期临床试验(TRuE-AD1 和 TRuE-AD2)中,局部用药 Ruxolitinib 也显示出抗炎和抗瘙痒作用。乌达帕替尼是一种高度特异性的 JAK1 抑制剂,曾在 RA 领域进行过探索,2019 年进一步获得 FDA 批准,用于治疗年龄≥12 岁的中重度难治性 AD 患者。269,270 ,271,272由于高达替尼对 JAK2 和 JAK3 的影响极小,因此副作用也很少,尤其是对造血系统的影响。几项三期试验(Measure Up 1、Measure Up 2 和 AD Up)表明,达帕替尼作为单药或与皮质类固醇外用药联用,对中重度 AD 患者具有良好的疗效和耐受性。包括 JADE MONO-1、MONO-2 和 TEEN 在内的 3 期研究表明,在早期缓解瘙痒、减少湿疹面积以及严重程度指数(EASI-75)和研究者总体评估(IGA)评分方面,200 或 100 毫克阿洛西替尼比安慰剂或杜匹单抗更有效。一项二期研究(NCT02201524)也表明,阿罗西替尼可使中重度银屑病患者的症状得到良好改善。289
肝细胞癌
多项研究表明,JAK-STAT 信号通路广泛参与多种类型的实体癌。肝细胞癌(HCC)是一种致命的异质性肿瘤,发病率和死亡率一直居高不下,每年导致 70 多万人死亡。尽管 HCC 的治疗方法有所改进,但 HCC 的临床预后仍然不容乐观,5 年生存率仅为 10%。肝癌的发生过程被认为涉及一系列基因改变和信号通路的异常变化。293,294,295,296,297人们已广泛研究了 JAK-STAT 信号通路在 HCC 发病过程中的潜在作用。使用干扰素-α(IFN-α)驱动宿主抗病毒反应是目前治疗慢性乙型肝炎的一线疗法,已被证实可延缓肝纤维化的进展,甚至延缓 HCC 的发生。作为一种重要的免疫相关细胞因子,IFN-α 可激活 JAK-STAT 信号转导,诱导多种具有抗病毒和免疫调节功能的 IFN 刺激基因。298Han M 等人发现,慢性乙型肝炎患者外周血中 STAT1、肌病毒抵抗(Mx)和 SOCS3 等 IFN 刺激基因的表达和激活与 IFN 诱导的抗病毒结果密切相关。301 ,302在产生 HBV 的 HepG2 细胞系中,乙肝病毒 X 蛋白通过使 STAT3 失活来削弱 IFN-α 信号传导,从而增加 SOCS3 和蛋白磷酸酶 2 A 的表达。Gao D 等人还观察到,miR-122 下调 SOCS3 可提高 IFN 抗 HBV 的有效性。303大量研究报告表明,SOCS3 在 HCC 组织中下调,并与 HCC 恶性转化有负面影响。304,305研究发现,SOCS3 甲基化在 HBV 阳性的 HCC 中比在正常肝组织中更常见,并与 HCC 的不良预后相关。Eyes absent homolog 2 可通过转录上调 SOCS3 的表达来阻断 JAK-STAT 信号转导,被认为是 HCC 中潜在的肿瘤抑制因子,并可抑制 HCC 的进展306。307此外,作为免疫检查点蛋白 B7 家族的成员,HHLA2 通过与 TMIGD2 结合被证明可激活 JAK-STAT 通路并加速 HCC 的进展。308
前列腺癌
雄激素受体(AR)靶向疗法(如恩扎鲁胺)主要通过攻击腺腔细胞,在前列腺癌患者身上取得了相当大的临床成功。不幸的是,晚期前列腺癌通常会转化为鳞状细胞癌或神经内分泌癌,从而产生耐药性。多项研究发现,前列腺癌中 JAK-STAT 信号的过度激活与癌系可塑性的调节、对 AR 靶向疗法的耐药性以及不良临床预后之间存在关联。311,312在小鼠器官组织、基因工程小鼠模型(GEMMs)和人类细胞系中,结合成纤维细胞生长因子受体(FGFR)信号传导,JAK-STAT 通路被证明可引发管腔型-基底型表型转换,进而形成阉割耐药前列腺癌(CRPC)31。这些结果为克服 CRPC 耐药性的新方法提供了重要的理论支持,并揭示了细胞系可塑性与癌变之间的关联。
乳腺癌
乳腺癌是全球妇女发病率最高的癌症。乳腺癌一般分为三类:激素受体阳性(HR+)、人表皮生长因子受体阳性(HER2+)和三阴性(TN)。313,314,315远处转移和耐药性是大多数乳腺癌患者预后不良的根本原因。316,317越来越多的证据表明,STAT 家族的所有成员都与乳腺癌密切相关,它们或具有促致癌特性,或具有抗致癌特性。318因此,乳腺癌为 JAK-STAT 在致癌过程中的双刃剑作用提供了一个范例。
319,320一些研究表明,STAT3 在乳腺癌细胞的增殖、侵袭、转移和免疫逃逸过程中发挥着核心作用。321,322此外,STAT3 还有助于维持乳腺癌干细胞(BCSCs)的表型和功能,这些细胞具有多能性、抗化疗和高自我更新能力。经典的 IL-6-JAK-STAT3 通路可上调与乳腺癌进展相关的基因,诱导对芳香化酶抑制剂 (AI) 化疗的耐受性,并与 HR+ 乳腺癌的低生存率相关。在 HER2+ 乳腺癌患者中,单独使用或与曲妥珠单抗联合使用选择性 JAK1/2 抑制剂鲁索利替尼(ruxolitinib)可抑制 IL-6-JAK2-STAT3-calprotectin 轴,从而削弱癌细胞的活力并改善临床疗效331。
尽管 JAK-STAT 信号具有促肿瘤作用,但一些临床前研究表明,JAK 抑制剂会损害 NK 细胞的抗肿瘤免疫力,并增加乳腺癌的转移负担。此外,JAK1 和 JAK2 抑制剂鲁索利替尼(ruxolitinib)可诱导巨噬细胞产生促炎介质,导致乳腺癌促肿瘤微环境的建立和耐药性的产生。333JAK-STAT 通路在乳腺癌中的多种作用凸显了 JAK-STAT 信号的重要性,并为有效治疗策略的开发提供了启示。
肾脏疾病
除 RA、血液恶性肿瘤和实体瘤外,转录组分析还发现,STAT1 和 STAT3 在肾小球和肾小管间质切片中的表达增加与局灶节段性肾小球硬化症的疾病进展有关334。Pang 等人证实,STAT3 激活介导了单侧输尿管梗阻模型中的肾脏纤维化,应用新型 STAT3 抑制剂 S3I-201 可抑制肾间质成纤维细胞的激活和纤维化。相反,Koike, K.等人观察到,在单侧输尿管阻塞模型中,激活的 JAK-STAT3 信号诱导近端肾小管细胞表达基质金属蛋白酶-2,从而缓解肾脏纤维化并促进组织修复。336有关 JAK-STAT 信号在肾组织修复和纤维化中功能的研究结果相互矛盾,这可能是由于动物模型和调控 JAK-STAT 通路的干预措施不同所致。
高尿酸血症诱导肾小管和肾间质细胞中的STAT3活化,伴随着肾脏纤维化和功能障碍,STAT3抑制剂S3I-201被证实能抑制JAK-STAT通路,达到抗纤维化的效果。在对肾小管间质纤维化的研究中,基于 miR-150 的 RNA 干扰被证实能逆转 SOCS1-JAK-STAT 通路的激活,缓解肾小管间质纤维化。337此外,在 IgA 肾病中,STAT1 和 STAT3 的过度活跃表达以及激活的 JAK-STAT 信号传导被证实伴随着进行性肾炎和蛋白尿。
研究还发现,JAK-STAT 信号的激活通过刺激肾小球系膜细胞过度生长,在糖尿病引起的肾损害中发挥着重要的致病作用。343在 STZ 诱导的糖尿病大鼠模型中,高血糖通过血管紧张素 II 触发肾小球系膜细胞中的 JAK-STAT 通路,血管紧张素 II 阻断治疗可逆转病理变化,改善肾功能。
常染色体显性多囊肾(ADPKD)是最常见的先天性肾脏疾病,通常伴有 JAK-STAT 通路的异常活性344,345。ADPKD 中 STAT5 的强迫表达会以生长激素依赖的方式转录上调细胞周期蛋白 D1,从而导致异常增殖。Pkd1nl/nl小鼠模型揭示了 JAK2 在囊肿衬里细胞和间质中的异位表达,并验证了抑制 JAK2 可延缓 ADPKD 的囊肿生长346。
使用 MRL/lpr 小鼠模型进行的实验表明,肾脏 CD8+组织驻留记忆 T 细胞的自我更新和效应功能需要 JAK/STAT 信号传导,这与狼疮肾炎的活动有关。STAT3 水平在狼疮肾炎中也急剧升高,并与不利的临床参数有关。347在非人灵长类异体移植物模型中,基于不含钙神经抑制剂的方案,JAK3 抑制剂 CP-690,550 与霉酚酸酯联合使用可降低肾急性排斥反应的发生率,并延长异体移植物的存活时间。此外,在肾细胞癌细胞中,IFN-γ 触发的 JAK-STAT 信号转导被 FGFR 通路抑制,使用 FGFR 受体酪氨酸激酶靶向抑制剂 lenvatinib 治疗可恢复抗肿瘤免疫力并增强免疫检查点抑制剂的疗效351。
JAK-STAT 通路与治疗
JAK-STAT 信号通路是细胞内的重要途径,各种细胞外可溶性分子通过它与膜受体结合,并将信号传递到细胞核。鉴于其在一系列癌症和自身免疫性疾病中的主要作用,JAK-STAT 信号通路已成为药物开发的一个重要靶点。针对 JAK-STAT 通路的药物可根据其影响信号转导过程的方式分为三类:细胞因子或受体抗体、STAT 抑制剂和 JAK 抑制剂。
上游细胞因子和受体是调节 JAK-STAT 信号通路功能的关键。355,356,357,358操纵 JAK-STAT 依赖性细胞因子和受体的药物(如用于经典阻断 IL-6 的 siltuximab 和 tocilizumab)可抑制 JAK-STAT 信号转导,因此已被用作许多疾病的治疗干预措施。
大多数 STAT 抑制剂通过限制 STAT 磷酸化、抑制 SH2- 介导的二聚化或诱导 STAT 降解发挥作用。鉴于活化的 STAT3 和 STAT5 在信号转导和致病过程中的重要功能,多种靶向 STAT3 和 STAT5 的抑制剂,包括多肽、多肽仿生学、寡核苷酸、siRNA、小分子和金属基复合物,已显示出良好的临床前疗效。数百篇研究文章介绍了 STAT 在疾病进展中的各种基本机制,以及数十种 STAT 抑制剂在临床前和临床试验中的疗效。
STAT3 是研究最广泛的 STAT 蛋白,在多种癌症中经常发生突变和过度激活,并促进癌细胞增殖、侵袭、转移和免疫逃避。318纳帕布卡辛(BBI608)是第一个进入三期临床试验的 STAT3 直接抑制剂,也是第一种癌症干性抑制剂,已被证明可抑制多种癌症中的各种恶性过程。此外,在顺铂耐药的 NSCLC 模型中,研究发现萘普卡西能降低关键干性相关基因的表达,消耗癌症干细胞(CSC)群,并使化疗耐药细胞对顺铂重新敏感375。此外,萘普卡西在恶性黑色素瘤中消除了小鼠和人类髓源性抑制细胞(MDSCs)的免疫抑制功能,并发现这与患者的临床预后有关。377最近一项关于萘普卡西单药治疗晚期结直肠癌的三期试验(NCT01830621)显示,萘普卡西组在患者预后方面的潜在疗效优于安慰剂组。 新型 STAT3 抑制剂 OPB-31121 可诱导细胞凋亡,并在胃癌细胞和异种移植模型中与 5-氟尿嘧啶和顺铂发挥协同作用。381,382在首次人体 I 期临床试验(NCT01184807)中,OPB-51602 在难治性实体瘤(尤其是 NSCLC)患者中显示出良好的抗肿瘤活性。此外,另一种 STAT3 抑制剂 TTI-101 (C188-9)也被发现可减轻 STAT3 SH2 结构域内磷酪氨酸(pY)肽结合位点的活化,而不影响线粒体功能。384在动物模型中进行的多项研究表明,该药对一系列自身免疫性疾病和癌症具有有效的治疗潜力和良好的安全性,这些疾病包括 SSc、克罗恩病、头颈部癌症以及肺癌、乳腺癌、结直肠癌和肝癌。385,386,387,388,389最近,TTI-101 被 FDA 授予 "快速通道 "称号,用于治疗复发/难治性局部晚期、不可切除或转移性肝细胞癌。TTI-101正在进行的一期临床试验(NCT03195699)显示,晚期实体瘤患者对TTI-101的耐受性良好。
此外,新的研究揭示了 STAT5 抑制剂的潜力。390STAT5抑制剂cllinflamozyde在美国食品药品管理局(FDA)最近批准的一项COVID-19临床试验中的初步结果显示,cllinflamozyde选择性地抑制了STAT5-TH-GM通路,从而缓解了严重的细胞因子风暴,并显著改善了COVID-19患者的临床预后。
此外,越来越多的策略将 STAT3 与免疫检查点抑制剂(如抗 CTLA-4 和 PD-1/PD-L1 抗体)、CAR-T 细胞疗法(NCT02906371)、干扰素基因刺激剂(STING)激动剂甚至癌症疫苗结合起来,在各种临床前试验中显示出令人鼓舞的协同抗肿瘤疗效。
根据以往的探索,STAT 抑制剂在抗肿瘤效果方面表现出了良好的价值。遗憾的是,大多数 STAT 抑制剂仍处于临床前开发阶段,由于大多数候选药物缺乏内在催化活性和选择性,很少有获准临床应用的。目前正在进行大量针对 STAT 的单一疗法和联合疗法的临床试验,为疾病治疗提供了一个极具吸引力的药物靶点。
尽管如此,天然产物及其衍生物已成为基于 JAK-STAT 通路的新型抗肿瘤药物的来源。395,396,397过去几十年来,体外和体内实验表明,各种天然产物对 STAT3 信号通路具有抑制作用,并表现出良好的抗癌活性。例如,植物化学物质姜黄素被证明能有效抑制 STAT3,并在乳腺癌、卵巢癌、肺癌和食管鳞状细胞癌中发挥抗癌作用。此外,研究还发现天然类黄酮 myricetin 可通过抑制肺癌 A549、乳腺癌 MDA-MB-231 和结肠癌 HCT116 癌细胞中 IFN-γ 激活的 JAK-STAT-IRF1 轴,下调程序性死亡配体-1(PD-L1)和吲哚胺 2,3-二氧 化酶 1(IDO1)的水平,恢复 T 细胞活性和抗肿瘤免疫力。Runtsch 等人提出,天然代谢物伊它康酸及其衍生物可通过阻断 JAK1-STAT6 通路磷酸化抑制 M2 极化,为 M2 巨噬细胞驱动的疾病提供了新的视角。404研究发现,作为一种新型天然 STAT3 抑制剂,XYA-2 可结合 STAT3 的 SH2 结构域,协同下调人胃癌细胞系和患者异种移植(PDX)小鼠模型中 MYC 和 SLC39A10 的水平,从而发挥抗癌作用。
此外,SOCS 蛋白是 JAK-STAT 通路负反馈环的一部分,以 SOCS 相互作用体为靶标的肽也有望成为疾病进展的抑制剂。
最近,几种 JAK 抑制剂已被临床批准用于治疗各种疾病,越来越多的创新候选药物正在进行临床前和临床试验(表3)。在此,我们回顾了主要 JAK 抑制剂在人类疾病中的关键应用和不良反应。
表 3 主要 JAK 抑制剂的应用和安全性
JAK 抑制剂是一种小分子抑制剂,可导致免疫抑制,减少由 JAK-STAT 信号驱动的病理性促炎细胞因子的产生,并抑制功能增益型 JAK 突变体。408,409目前正在进行各种 JAK 抑制剂的临床前和临床研究,以治疗一系列自身免疫性疾病和癌症。
第一代口服 JAK 抑制剂彻底改变了一组异质性疾病的治疗方法。418第一个获得临床批准的 JAK 抑制剂是 JAK1 和JAK2抑制剂 ruxolitinib,它于 2011 年获得美国食品药品管理局 (FDA) 批准用于治疗骨髓纤维化。Verstovsek, S.等人进行的 COMFORT-I 试验表明,Ruxolitinib 对中-2 级或高风险骨髓纤维化患者有临床益处;与安慰剂组相比,Ruxolitinib 组患者的表现有所改善,脾脏体积缩小,总生存期延长。后来,COMFORT-II 试验表明,与接受现有最佳疗法的患者相比,骨髓纤维化患者可从持续的 Ruxolitinib 治疗中获益,突出了 Ruxolitinib 的长期有效性和总生存率优势。Kvasnicka,H. M. 等人报告说,长期服用 48 个月或 60 个月的 ruxolitinib 可改善和稳定骨髓纤维化的进展。427Zeiser 等人进行了一项 3 期试验,评估 ruxolitinib 对糖皮质激素难治性或依赖性慢性移植物抗宿主病患者的疗效。 结果显示,与接受普通二线疗法的患者相比,165 名接受 ruxolitinib 治疗的患者在 24 周时的总体反应更佳,12 个月时的反应持续时间更长,无失败生存率更高。428在一项针对炎症性肠病患者的单中心回顾性研究中,6 个月的 ruxolitinib 治疗可改善肠外症状、大便次数、类固醇减量和营养状况。430最近,RUXCOVID 3 期试验将 432 名患者随机分为 Ruxolitinib 组(n= 287)和安慰剂组(n= 145)加标准护理,以评估 Ruxolitinib 治疗 2019 年冠状病毒病(COVID-19)的疗效。结果显示,与接受安慰剂治疗的患者相比,接受 Ruxolitinib 治疗的患者的中位康复时间缩短了 1 天。431然而,Fisher, D. 等人最近指出,骨髓纤维化患者在接受 Ruxolitinib 治疗后仍会出现多细胞因子过度分泌的情况。432值得注意的是,据报道,一名 75 岁的骨髓纤维化患者在接受 Ruxolitinib 治疗后出现了进行性多灶性白质脑病。433还有几项研究报告称,快速停用 ruxolitinib 会诱发危及生命的停药症状,这可能是由于炎症细胞因子的活性发生了剧烈变化。434,435,436,437,438,439,440,441,442
具有优先选择性的JAK3和 JAK1 抑制剂托法替尼是首个获准用于对甲氨蝶呤等传统药物反应不佳的 RA 患者的 JAK 抑制剂。多项研究表明,与其他 DMARDs 相比,托法替尼对 RA 患者的疗效更佳,安全性更高。460,461,462,463,464,465,466,467Sandborn, W. J.等人进行了三项三期试验,证实了托法替尼作为中度至重度活动性溃疡性结肠炎患者的诱导和维持治疗具有良好疗效。 468在一项涉及 10 名肉样瘤病患者的开放标签试验中,托法替尼主要通过抑制 1 型免疫改善了所有 10 名患者的皮肤和内脏器官症状469。You, H. 等人的研究表明,在改善弥漫性皮肤系统性硬化症难治性皮肤增厚患者的改良罗德南皮肤评分方面,托法替尼与传统的免疫抑制剂效果相同或更好。P.S.Changelian等人通过在小鼠心脏移植模型和非人灵长类肾移植模型中使用托法替尼介导的免疫抑制来预防器官排斥反应,证明了托法替尼对预防体内异体移植排斥反应的潜在益处。为了进一步探讨托法替尼在现实世界中的安全性,Khosrow-Khavar, F.等人招募了两组接受托法替尼或肿瘤坏死因子(TNF)抑制剂治疗的RA患者,结果发现托法替尼治疗后患者心血管不良反应的增加在统计学上并不显著。然而,口服监测报告显示,与肿瘤坏死因子抑制剂相比,在年龄超过 50 岁且至少有一个额外心血管风险因素的 RA 患者中,托法替尼导致感染和心血管事件的发生率更高。 在临床前试验和临床前基因研究中,发现 ruxolitinib 具有潜在的心脏毒性,可能表现为毒性心肌损伤、左室收缩功能下降或心力衰竭。472,473,474
选择性JAK1和JAK2抑制剂巴利昔尼于2017年获批用于治疗RA,并被广泛证实可改善对甲氨蝶呤反应不充分的RA患者的临床症状。RA-BEAM试验涉及对甲氨蝶呤反应不充分的中度至重度活动性RA患者,结果显示,在体征和症状、身体功能以及关节结构损伤方面,巴利昔尼优于安慰剂。485,486,487,488,489,490RA-BEACON试验探讨了巴利替尼对bDMARDs难治的RA患者的疗效。在两项针对重度斑秃患者的三期试验中,巴利昔替尼在促进毛发再生方面优于安慰剂。研究还发现,巴利替尼对麻木相关激酶(NAK)有很高的亲和力,这使其能够抑制 NAK 的活性,阻断凝胶酶介导的内吞作用,从而减少 COVID-19 病毒感染。491,492此外,一项试验性研究显示,巴利昔尼改善了 12 名轻中度 COVID-19 肺炎患者的呼吸道症状,且不会导致严重不良事件。Cantini, F.等人还对 113 名连续住院的中度肺炎患者进行了回顾性多中心研究,验证了巴利昔尼在降低病毒负荷、死亡率和住院率方面的有益作用。494正在进行的 RECOVERY 试验进一步证实了巴利昔尼在降低 COVID-19 死亡率方面的有效性。一项针对 3492 名接受巴利昔尼治疗的 RA 患者的综合研究发现,使用巴利昔尼与主要不良心血管事件、动脉血栓事件或充血性心力衰竭之间没有相关性。495
496大量 2b 期试验(如 RAJ1、RA21 和 RA22)表明,peficitinib 作为一种单一疗法或与 csDMARDs 联用,可在中重度 RA 患者中实现 12 周内快速且具有统计学意义的美国风湿病学会(ACR)应答率。3 期试验(RAJ3 和 RAJ4)也观察到,治疗 12 周后,peficitinib 在减轻 RA 症状和关节破坏方面表现更佳。此外,一项 2b 期试验显示,溃疡性结肠炎患者在接受≥75 毫克培非替尼治疗后,第 8 周的临床反应率、缓解率和粘膜愈合率均有所提高194。根据一项 2a 期多中心安慰剂对照研究的结果,培非替尼对中重度银屑病患者的临床和组织学病变也有剂量依赖性改善193。此外,peficitinib 的安全性也是可以接受的,通常包括鼻咽炎、带状疱疹、腹泻、血肌酸磷酸激酶水平升高和淋巴细胞减少。497Delgocitinib 是一种泛 JAK 抑制剂,对促炎细胞因子具有广泛的抑制作用,对多种炎症性皮肤病有效,包括 AD、湿疹、盘状红斑狼疮、银屑病和斑秃。498,499,500在三期试验中,中度至重度注意力缺失症患者局部服用地高替尼后,疗效终点得到了有临床意义的改善,地高替尼因此被批准用于注意力缺失症的治疗。此外,在对儿童和成人注意力缺失症患者进行的整个研究期间,大多数不良反应都很轻微,与使用delgocitinib无关。501,502,503,504
第一代 JAK 抑制剂被认为是针对多种 JAK 异构体的泛 JAK 抑制剂。鉴于其高度保守的 ATP 结合位点,JAK 抑制剂可产生广泛的作用,但也会引起副作用,包括心血管事件、血栓栓塞事件、感染和致癌并发症505。因此,开发更具选择性的 JAK 抑制剂,强调增强特异性和减少不良反应已成为药物研究的重点506。507,508,509,510,511美国国家综合治疗网络也将费拉替尼列为一类推荐药物,用于治疗血小板计数≥50×109/L的骨髓纤维化高危患者512。随机的 JAKARTA 3 期试验和非随机的 JAKARTA-2 2 期试验一致显示,对于鲁索利替尼耐药或不耐受的中度-1、中度-2 或高风险骨髓纤维化患者,非瑞替尼可减轻症状和脾脏肿大。此外,Harrison, C. N.等人进行了一项 2 期多中心研究,报告称约 30% 对鲁索利替尼耐药或不耐受的骨髓纤维化患者接受 400 毫克非瑞替尼治疗后,脾脏体积和临床症状显著减少。417然而,据报道,在 JAKARTA 试验中,670 例接受非瑞替尼治疗的患者中有 8 例和 4 例接受 500 毫克/天非瑞替尼治疗的女性患者出现了经磁共振成像证实的韦尼克氏脑病,这引起了人们的关注,并阻碍了非瑞替尼的进一步临床开发510。513,514此前的 2a 和 2b 期试验均报告称,作为一种单一疗法或与甲氨蝶呤联用,地克诺替尼有助于改善对甲氨蝶呤反应不足的 RA 患者的体征和症状。在这些试验中发现的主要不良反应包括头痛、恶心、转氨酶、脂蛋白和肌酐水平升高以及淋巴细胞和中性粒细胞数量减少。迄今为止,伊塔替尼主要用于治疗移植物抗宿主病(GVHD)。 最近,一项多中心 2 期试验(NCT03846479)报告称,低风险急性 GVHD 患者在不常规使用全身性皮质类固醇(SCS)的情况下服用 28 天的伊塔替尼,可获得有效的药物反应率并减少症状性复发518。一项 3 期试验(GRAVITAS-301,NCT03139604)表明,伊塔替尼与皮质类固醇联合治疗急性 GVHD 患者第 28 天的总体应答率 (ORR) 并未达到预期改善效果。欧洲血液和骨髓移植学会(EBMT 2022)报告了一项 2 期试验(NCT 04071366)的初步结果,指出伊塔替尼在预防免疫疗法后的细胞因子释放综合征(CRS)方面表现良好。此外,在世界肺癌大会(WCLC 2022)上,PD-L1高表达(≥50%)的NSCLC患者在接受伊塔替尼和pembrolizumab联合治疗6周后,疗效和耐受性均表现良好。最常见的不良反应主要包括血小板和中性粒细胞计数下降、贫血和高血糖。要进一步了解伊沙替尼治疗急性GVHD的效果,还需要进一步的研究。
目前,正在探索几种处于不同临床试验阶段的 JAK 抑制剂,以扩大潜在适应症、提高药物依从性并实现更好的安全性。例如,upadacitinib(ABT-494)是一种强效的选择性 JAK1 抑制剂,已获得 FDA 批准,可用于中重度 RA、银屑病关节炎(PsA)、强直性脊柱炎(AS)和非放射性轴性脊柱关节炎(nr-axSpA)患者的临床治疗。520、521、522SELECT-MONOTHERAPY 研究发现,在中度至重度活动性 RA 患者中联合使用稳定的 csDMARDs 时,upadacitinib 单药治疗可改善临床症状523。在对一种或多种 TNF 抑制剂反应不充分或不耐受的 PsA 患者中开展的两项经典 3 期试验(SELECT-PsA 1 和 SELECT-PsA2)发现,与安慰剂或阿达木单抗相比,奥达帕替尼可减轻 PsA 疾病的严重程度。此外,在为期 14 周的治疗中,发现达帕替尼对 bDMARD 反应不充分的活动性 AS 患者有效。524最近,基于两项诱导研究(U-ACHIEVE 和 U-ACCOMPLISH)和一项维持研究(U-ACHIEVE)的积极临床结果,欧盟通过了针对炎症性肠病患者开发奥达帕替尼治疗的批准。525尽管如此,要验证达帕替尼的长期安全性,还需要进行更多具有大量人群样本的试验,因为具有类似机制的其他 JAK 抑制剂也报告了不良事件,如心血管事件、栓塞和血栓形成、癌症甚至死亡。
阿罗西替尼是另一种经 FDA 批准、正在开发用于 AD 治疗的 JAK1 选择性抑制剂,它通过阻断参与 AD 病理学的几种关键细胞因子(包括 IL-4、IL-13、IL-22 和 IL-31)发挥作用。在一系列三期试验(JADE Mono-1、MONO-2、TEEN、COMPARE、EXTEND和REGIMEN)中,阿洛西替尼单药治疗明显改善了瘙痒和AD疾病的严重程度,因此阿洛西替尼的适应症扩大到12-17岁患有中重度AD的青少年。531阿罗西替尼的安全性尚可,常见的不良反应包括恶心、头痛、痤疮和血小板计数减少。532,533因此,在将阿罗西替尼用于 AD 的长期治疗之前,需要对其益害比进行深入研究。引人注目的是,deucravacitinib(BMS-986165)通过产生独特的异构机制与 TYK2 调节域相结合,而不影响 JAK1/2/3 的功能,成为首个用于治疗银屑病且安全性良好的口服药物。534Deucravacitinib 也是首个获批用于治疗中重度斑块状银屑病的 TYK2 选择性抑制剂;它于 2022 年 9 月获得美国 FDA 批准,随后在日本获批用于治疗斑块状、泛发性脓疱型和红皮病型银屑病535。535两项关键的 3 期 POETYK PSO-1 和 POETYK PSO-2 试验证明,与安慰剂或阿普司特相比,在多个疗效终点方面,deucravacitinib 对中重度斑块状银屑病的疗效更优。deucravacitinib 最常见的不良反应包括轻度至中度鼻咽炎和上呼吸道感染。最近宣布的 POETYK PSO-3 试验首次拓宽了在亚洲人群中探索 deucravacitinib 疗效的范围。第30届欧洲皮肤病与性病学学会(EADV)大会的口头结果表明,对亚洲中重度斑块状银屑病患者服用deucravacitinib可获得持续疗效,甚至对难治性头皮银屑病也有良好疗效。与此同时,多个大型临床试验也在探索 deucravacitinib 对银屑病关节炎(PsA)、系统性红斑狼疮和炎症性肠病等多种免疫相关疾病的疗效和安全性。538
近年来,我国自主研发的多个JAK抑制剂(如贾克替尼、戈利昔替尼、依伐替尼、伊他替尼等)处于不同阶段的临床试验中,被认为具有巨大的临床潜力。2022 年 10 月,广泛抑制 JAK1、JAK2、JAK3、TYK2 和 ACVR1 的莫美罗替尼氚化物 Jaktinib 成为首个获批用于治疗中重度骨髓纤维化的国产 JAK 抑制剂。已发表的一项 2 期试验显示,贾克替尼(jaktinib)可缩小中重度骨髓纤维化患者的脾脏大小,并缓解其临床症状546。3 期试验(NCT04617028)进一步报告了 jaktinib 治疗骨髓纤维化的令人鼓舞的疗效,与羟基脲相比,症状、贫血和脾脏体积均有显著改善。根据现有的疗效和安全性报告,需要获得批准才能开展 Jaktinib 用于 COVID-19、AD、系统性红斑狼疮、斑秃和轻中度银屑病等一系列疾病的临床试验547。
总体而言,JAK 抑制剂作为靶向疗法前景广阔。然而,要充分探索其长期安全性、持久性和有效性,还需要足够的实际证据。
该领域当前的挑战和方向
对 JAK-STAT 信号转导的深入了解为新型药物的开发提供了许多启示。548,549,550然而,要提高 JAK-STAT 信号转导的靶点选择性仍面临许多挑战,而且仍需探索所开发药物的长期安全性。551,552ATP、JAK 酶和 JAK 抑制剂的 Michaelis 平衡表明,JAK 与其底物的结合受细胞内药物浓度和药物选择性的影响553。然而,药物浓度取决于多种因素,如患者的年龄、体重、肝肾功能和药物相互作用,因此要实现 JAK 抑制剂的绝对选择性一直是个挑战。目前正在开发更具选择性的 JAK 抑制剂,以减少其对细胞因子功能的不良影响,并进一步提高整体安全性和疗效。555,556,557例如,目前正在进一步开发 JAK 抑制剂阿洛西替尼(abrocitinib)和德尔戈西替尼(delgocitinib),以优化治疗效率,将脱靶效应降至最低。
558,559同时抑制 JAK-STAT 信号通路和其他潜在靶点似乎是另一种可行的药物开发方案。例如,fedratinib 是一种创新型 JAK 抑制剂,可同时靶向 JAK2 和 Fms 样酪氨酸激酶 3 (FLT3),对原始造血祖细胞的存活和增殖有重要影响,已被批准作为中度或高风险骨髓纤维化的口服治疗药物560,561,562。
JAK 抑制剂的局部或吸入应用是另一个令人兴奋的研究领域,目前正在进行多个动物模型和更大规模的临床试验。548,563肠道限制性泛 JAK 抑制剂 TD-1473 预计将很快上市,用于治疗炎症性肠病,与其他治疗方案相比,其全身不良副作用较少。
结论
作为一种主要的典型信号通路,JAK-STAT 信号通路将细胞因子激活的细胞外信号转导至细胞核,从而介导基因表达,在一系列细胞过程,尤其是具有免疫调节作用的过程中发挥着不可或缺的功能。JAK-STAT 信号的异常激活是许多疾病(尤其是免疫相关疾病和癌症)发生和发展的核心原因。针对 JAK-STAT 通路的药物已被美国食品及药物管理局批准作为某些疾病的替代治疗药物,并已显示出强大的临床疗效。目前有大量靶向 JAK-STAT 通路的新型药物正在研发中,但仍很少有证据表明选择性 JAK 抑制剂和泛选择性 JAK 抑制剂的疗效存在差异。在疾病早期阶段使用 JAK 抑制剂以及与其他常规药物联合使用的探索也在进行中。最近的研究表明,部分患者对 JAK 抑制剂无反应,并试图揭示相关的耐药机制。未来的研究应全面揭示 JAK-STAT 通路在疾病发展中的生理和致病机制,并旨在确定生物标志物,以评估 JAK 抑制剂的长期疗效和安全性,同时优化 JAK 抑制剂对不同阶段和不同病情严重程度患者的疗效,实现个体化治疗。总之,了解 JAK-STAT 通路与免疫调节和疾病进展的关系将为治疗各种疾病,尤其是免疫相关疾病和癌症提供新的治疗策略。
参考资料
Leonard, W. J. & O'Shea, J. J. Jaks and STATs: biological implications.Annu.Rev. Immunol.16, 293-322 (1998).
O'Shea, J. J. Jaks, STATs, cytokine signal transduction, and immunoregulation: are we there yet?Immunity7,1-11(1997 年)。
Hou, X. S. & Perrimon, N. The JAK-STAT pathway in drosophila.Trends Genet.13, 105-110 (1997).
Barrat, F. J., Crow, M. K. & Ivashkiv, L. B. Interferon target-gene expression and epigenomic signatures in health and disease.Nat.Immunol.20,1574-1583 (2019)。
Meraz, M. A. et al. 小鼠 Stat1 基因的靶向破坏揭示了 JAK-STAT 信号通路意想不到的生理特异性。Cell84, 431-442 (1996).Yamaoka, K. et al. The Janus kinases (Jaks). Genome Biol. 5, 253 (2004).
Harpur, A. G., Andres, A. C., Ziemiecki, A., Aston, R. R. & Wilks, A. F. JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene 7, 1347–1353 (1992).
Rane, S. G. & Reddy, E. P. JAK3: a novel JAK kinase associated with terminal differentiation of hematopoietic cells. Oncogene 9, 2415–2423 (1994).
Minegishi, Y. et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 25, 745–755 (2006).
Levy, D. E. & Darnell, J. E. Jr Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662 (2002).
Darnell, J. E. Jr. STATs and gene regulation. Science 277, 1630–1635 (1997).
Arimoto, K. I. et al. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat. Struct. Mol. Biol. 24, 279–289 (2017).
Chen, W. & Daines, M. O. Khurana Hershey, G. K. Turning off signal transducer and activator of transcription (STAT): the negative regulation of STAT signaling. J. Allergy Clin. Immunol. 114, 476–489 (2004). quiz 490.
Darnell, J. E. Jr Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2, 740–749 (2002).
McBride, K. M., Banninger, G., McDonald, C. & Reich, N. C. Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-alpha. EMBO J. 21, 1754–1763 (2002).
O’Shea, J. J., Gadina, M. & Schreiber, R. D. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell 109, S121–S131 (2002).
Ihle, J. N. & Kerr, I. M. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 11, 69–74 (1995).
Ihle, J. N. et al. Signaling by the cytokine receptor superfamily: JAKs and STATs. Trends Biochem. Sci. 19, 222–227 (1994).
Koskela, H. L. et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 366, 1905–1913 (2012).
Jerez, A. et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood 120, 3048–3057 (2012).
Constantinescu, S. N., Girardot, M. & Pecquet, C. Mining for JAK-STAT mutations in cancer. Trends Biochem. Sci. 33, 122–131 (2008).
Mirtti, T. et al. Nuclear Stat5a/b predicts early recurrence and prostate cancer-specific death in patients treated by radical prostatectomy. Hum. Pathol. 44, 310–319 (2013).
Owen, K. L., Brockwell, N. K., Parker, B. S. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers (Basel). 11, 2002 (2019).
Meissl, K., Macho-Maschler, S., Müller, M. & Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 89, 12–20 (2017).
Spiotto, M. T. & Chung, T. D. STAT3 mediates IL-6-induced neuroendocrine differentiation in prostate cancer cells. Prostate 42, 186–195 (2000).
Rojas, A. et al. IL-6 promotes prostate tumorigenesis and progression through autocrine cross-activation of IGF-IR. Oncogene 30, 2345–2355 (2011).
Lee, S. O. et al. RNA interference targeting Stat3 inhibits growth and induces apoptosis of human prostate cancer cells. Prostate 60, 303–309 (2004).
Tzeng, H. T., Chyuan, I. T. & Lai, J. H. Targeting the JAK-STAT pathway in autoimmune diseases and cancers: a focus on molecular mechanisms and therapeutic potential. Biochem. Pharm. 193, 114760 (2021).
Lai, S. Y. & Johnson, F. M. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resist. Updat. 13, 67–78 (2010).
Burmester, G. R. et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet 381, 451–460 (2013).
Chan, J. M. et al. Lineage plasticity in prostate cancer depends on JAK/STAT inflammatory signaling. Science 377, 1180–1191 (2022).
Boyle, D. L. et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann. Rheum. Dis. 74, 1311–1316 (2015).
Palmroth, M. et al. Tofacitinib suppresses several JAK-STAT pathways in rheumatoid arthritis in vivo and baseline signaling profile associates with treatment response. Front. Immunol. 12, 738481 (2021).
Tucci, G. et al. Baricitinib therapy response in rheumatoid arthritis patients associates to STAT1 phosphorylation in monocytes. Front. Immunol. 13, 932240 (2022).
Verstovsek, S. et al. A phase I, open-label, dose-escalation, multicenter study of the JAK2 inhibitor NS-018 in patients with myelofibrosis. Leukemia 31, 393–402 (2017).
Benucci, M. et al. Cardiovascular safety, cancer and Jak-inhibitors: differences to be highlighted. Pharm. Res. 183, 106359 (2022).
Desai, R. J., Pawar, A., Weinblatt, M. E. & Kim, S. C. Comparative risk of venous thromboembolism in rheumatoid arthritis patients receiving tofacitinib versus those receiving tumor necrosis factor inhibitors: an observational cohort study. Arthritis Rheumatol. 71, 892–900 (2019).
O’Shea, J. J. et al. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu. Rev. Med. 66, 311–328 (2015).
Singh, S. R., Chen, X. & Hou, S. X. JAK/STAT signaling regulates tissue outgrowth and male germline stem cell fate in Drosophila. Cell Res. 15, 1–5 (2005).
Fahmideh, H. et al. The role of natural products as inhibitors of JAK/STAT signaling pathways in glioblastoma treatment. Oxid. Med Cell Longev. 2022, 7838583 (2022).
Gordon, M. J., Smith, M. R., Nastoupil, L. J. Follicular lymphoma: the long and winding road leading to your cure? Blood Rev. 57, 100992 (2022).
Banerjee, S., Biehl, A., Gadina, M., Hasni, S. & Schwartz, D. M. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs 77, 521–546 (2017).
Villarino, A. V., Kanno, Y. & O’Shea, J. J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 18, 374–384 (2017).
Waldmann, T. A. & Chen, J. Disorders of the JAK/STAT pathway in T cell lymphoma pathogenesis: implications for immunotherapy. Annu. Rev. Immunol. 35, 533–550 (2017).
Roskoski, R. Jr Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharm. Res. 111, 784–803 (2016).
Yin, T., Yang, L. & Yang, Y. C. Tyrosine phosphorylation and activation of JAK family tyrosine kinases by interleukin-9 in MO7E cells. Blood 85, 3101–3106 (1995).
Choudhury, G. G., Marra, F., Kiyomoto, H. & Abboud, H. E. PDGF stimulates tyrosine phosphorylation of JAK 1 protein tyrosine kinase in human mesangial cells. Kidney Int. 49, 19–25 (1996).
Miyazaki, T. et al. Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Science 266, 1045–1047 (1994).
Russell, S. M. et al. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID. Science 266, 1042–1045 (1994).
Saijo, K., Park, S. Y., Ishida, Y., Arase, H. & Saito, T. Crucial role of Jak3 in negative selection of self-reactive T cells. J. Exp. Med. 185, 351–356 (1997).
Das Gupta, D. et al. IRF4 deficiency vulnerates B-cell progeny for leukemogenesis via somatically acquired Jak3 mutations conferring IL-7 hypersensitivity. Cell Death Differ. 29, 2163–2176 (2022).
Liao, W. et al. Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression. Proc. Natl. Acad. Sci. USA 111, 3508–3513 (2014).
Malka, Y. et al. Ligand-independent homomeric and heteromeric complexes between interleukin-2 or -9 receptor subunits and the gamma chain. J. Biol. Chem. 283, 33569–33577 (2008).
Degryse, S. et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood 124, 3092–3100 (2014).
Barcia Durán, J. G. et al. Endothelial Jak3 expression enhances pro-hematopoietic angiocrine function in mice. Commun. Biol. 4, 406 (2021).
Glassman, C. R. et al. Structure of a Janus kinase cytokine receptor complex reveals the basis for dimeric activation. Science 376, 163–169 (2022).
Ferrao, R. & Lupardus, P. J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK-Receptor Interactions. Front. Endocrinol. (Lausanne) 8, 71 (2017).
Zhou, Y. J. et al. Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases. Mol. Cell 8, 959–969 (2001).
Goult, B. T. et al. Structure of a double ubiquitin-like domain in the talin head: a role in integrin activation. EMBO J. 29, 1069–1080 (2010).
Lupardus, P. J. et al. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc. Natl Acad. Sci. USA 111, 8025–8030 (2014).
Bandaranayake, R. M. et al. Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat. Struct. Mol. Biol. 19, 754–759 (2012).
Barahmand-Pour, F., Meinke, A., Groner, B. & Decker, T. Jak2-Stat5 interactions analyzed in yeast. J. Biol. Chem. 273, 12567–12575 (1998).
Guschin, D. et al. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin-6. EMBO J. 14, 1421–1429 (1995).
Finbloom, D. S. & Winestock, K. D. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J. Immunol. 155, 1079–1090 (1995).
Yang, H. et al. The influenza virus PB2 protein evades antiviral innate immunity by inhibiting JAK1/STAT signalling. Nat. Commun. 13, 6288 (2022).
Shao, T. et al. Treatment with a JAK1/2 inhibitor ameliorates murine autoimmune cholangitis induced by IFN overexpression. Cell Mol. Immunol. 19, 1130–1140 (2022).
Gerlach, K. et al. The JAK1/3 inhibitor tofacitinib suppresses T cell homing and activation in chronic intestinal inflammation. J. Crohns Colitis 18, jjaa162 (2020).
Lee, T. S. et al. Mechanisms of constitutive activation of Janus kinase 2-V617F revealed at the atomic level through molecular dynamics simulations. Cancer 115, 1692–1700 (2009).
Roskoski, R. Jr Janus kinase (JAK) inhibitors in the treatment of neoplastic and inflammatory disorders. Pharm. Res. 183, 106362 (2022).
Liosi, M. E. et al. Insights on JAK2 modulation by potent, selective, and cell-permeable pseudokinase-domain ligands. J. Med. Chem. 65, 8380–8400 (2022).
Rai, S. et al. Inhibition of interleukin-1β reduces myelofibrosis and osteosclerosis in mice with JAK2-V617F driven myeloproliferative neoplasm. Nat. Commun. 13, 5346 (2022).
Rahman, M. F. et al. Interleukin-1 contributes to clonal expansion and progression of bone marrow fibrosis in JAK2V617F-induced myeloproliferative neoplasm. Nat. Commun. 13, 5347 (2022).
Neubauer, H. et al. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 93, 397–409 (1998).
Mu, W., Ao, J., Li, Y., Zhang, J. & Duan, C. Exploring the protective mechanisms of total tannins from Geum japonicum var. chinense F.Bolle in mice with hematopoietic dysfunction via the JAK2/STAT3/5 signaling pathway. J. Ethnopharmacol. 296, 115507 (2022).
Rubio, T., Viana, R., Moreno-Estellés, M., Campos-Rodríguez, Á. & Sanz, P. TNF and IL6/Jak2 signaling pathways are the main contributors of the glia-derived neuroinflammation present in Lafora disease, a fatal form of progressive myoclonus epilepsy. Neurobiol. Dis. 176, 105964 (2023).
Hu, X., Li, J., Fu, M., Zhao, X. & Wang, W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct. Target Ther. 6, 402 (2021).
Dodington, D. W., Desai, H. R. & Woo, M. JAK/STAT - emerging players in metabolism. Trends Endocrinol. Metab. 29, 55–65 (2018).
DiSanto, J. P. Cytokines: shared receptors, distinct functions. Curr. Biol. 7, R424–R426 (1997).
Schindler, C. & Darnell, J. E. Jr Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu. Rev. Biochem. 64, 621–651 (1995).
Ihle, J. N., Witthuhn, B. A., Quelle, F. W., Yamamoto, K. & Silvennoinen, O. Signaling through the hematopoietic cytokine receptors. Annu. Rev. Immunol. 13, 369–398 (1995).
Thomis, D. C., Gurniak, C. B., Tivol, E., Sharpe, A. H. & Berg, L. J. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science 270, 794–797 (1995).
Karaghiosoff, M. et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity 13, 549–560 (2000).
Rusiñol, L. & Puig, L. Tyk2 targeting in immune-mediated inflammatory diseases. Int J. Mol. Sci. 24, 3391 (2023).
Liau, N. P. D. et al. Enzymatic characterization of wild-type and mutant janus kinase 1. Cancers (Basel) 11, 1701 (2019).
Gauzzi, M. C. et al. The amino-terminal region of Tyk2 sustains the level of interferon alpha receptor 1, a component of the interferon alpha/beta receptor. Proc. Natl Acad. Sci. USA 94, 11839–11844 (1997).
Velazquez, L. et al. Distinct domains of the protein tyrosine kinase tyk2 required for binding of interferon-alpha/beta and for signal transduction. J. Biol. Chem. 270, 3327–3334 (1995).
Stahl, N. et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 263, 92–95 (1994).
Bacon, C. M. et al. Interleukin 12 (IL-12) induces tyrosine phosphorylation of JAK2 and TYK2: differential use of Janus family tyrosine kinases by IL-2 and IL-12. J. Exp. Med. 181, 399–404 (1995).
Welham, M. J., Learmonth, L., Bone, H. & Schrader, J. W. Interleukin-13 signal transduction in lymphohemopoietic cells. Similarities and differences in signal transduction with interleukin-4 and insulin. J. Biol. Chem. 270, 12286–12296 (1995).
Watford, W. T. & O’Shea, J. J. Human tyk2 kinase deficiency: another primary immunodeficiency syndrome. Immunity 25, 695–697 (2006).
Verhoeven, Y. et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 60, 41–56 (2020).
Furqan, M. et al. STAT inhibitors for cancer therapy. J. Hematol. Oncol. 6, 90 (2013).
Dorritie, K. A., McCubrey, J. A. & Johnson, D. E. STAT transcription factors in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia 28, 248–257 (2014).
Zhang, T., Kee, W. H., Seow, K. T., Fung, W. & Cao, X. The coiled-coil domain of Stat3 is essential for its SH2 domain-mediated receptor binding and subsequent activation induced by epidermal growth factor and interleukin-6. Mol. Cell Biol. 20, 7132–7139 (2000).
Kwock, J. T. et al. IL-27 signaling activates skin cells to induce innate antiviral proteins and protects against Zika virus infection. Sci. Adv. 6, eaay3245 (2020).
Léger, T., Balaguer, P., Le Hégarat, L. & Fessard, V. Fate and PPARγ and STATs-driven effects of the mitochondrial complex I inhibitor tebufenpyrad in liver cells revealed with multi-omics. J. Hazard Mater. 442, 130083 (2023).
Lu, C. et al. IFNGR/STAT1 signaling in recipient hematopoietic antigen presenting cells suppresses graft-versus-host disease. J. Clin. Invest. 133, e125986 (2022).
Wang, J. et al. Leukemia inhibitory factor protects against graft-versus-host disease while preserving graft-versus-leukemia activity. Blood 140, 2076–2090 (2022).
Qin, Y. et al. Age-associated B cells contribute to the pathogenesis of rheumatoid arthritis by inducing activation of fibroblast-like synoviocytes via TNF-α-mediated ERK1/2 and JAK-STAT1 pathways. Ann. Rheum. Dis. 81, 1504–1514 (2022).
Pelham, S. J. et al. STAT5B restrains human B-cell differentiation to maintain humoral immune homeostasis. J. Allergy Clin. Immunol. 150, 931–946 (2022).
Duetsch, G. et al. STAT6 as an asthma candidate gene: polymorphism-screening, association and haplotype analysis in a Caucasian sib-pair study. Hum. Mol. Genet. 11, 613–621 (2002).
Schindler, C., Levy, D. E. & Decker, T. JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282, 20059–20063 (2007).
Chen, H. et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 147, 436–446 (2011).
Drube, S. et al. Interleukin-3 stabilizes CD124/IL-4α surface expression in mast cells via Tyk2 and STAT6. Immunology. 169, 102–112 (2022).
Hoey, T. & Schindler, U. STAT structure and function in signaling. Curr. Opin. Genet. Dev. 8, 582–587 (1998).
Shuai, K. The STAT family of proteins in cytokine signaling. Prog. Biophys. Mol. Biol. 71, 405–422 (1999).
Li, W. X. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 18, 545–551 (2008).
Baldini, C., Moriconi, F. R., Galimberti, S., Libby, P. & De Caterina, R. The JAK-STAT pathway: an emerging target for cardiovascular disease in rheumatoid arthritis and myeloproliferative neoplasms. Eur. Heart J. 42, 4389–4400 (2021).
Xin, P. et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 80, 106210 (2020).
Hong, J. et al. PRC2-mediated epigenetic suppression of Type I IFN-STAT2 signaling impairs antitumor immunity in luminal breast cancer. Cancer Res. 82, 4624–4640 (2022).
Gothe, F. et al. Aberrant inflammatory responses to type I interferon in STAT2 or IRF9 deficiency. J. Allergy Clin. Immunol. 150, 955–964.e916 (2022).
Li, Y. J., Zhang, C., Martincuks, A., Herrmann, A. & Yu, H. STAT proteins in cancer: orchestration of metabolism. Nat. Rev. Cancer 23, 115–134 (2023).
Shih, P. C. The role of the STAT3 signaling transduction pathways in radioresistance. Pharm. Ther. 234, 108118 (2022).
Zhang, L. et al. Signal transducer and activator of transcription 3 signaling in tumor immune evasion. Pharm. Ther. 230, 107969 (2022).
Yoshimura, A. & Yasukawa, H. JAK’s SOCS: a mechanism of inhibition. Immunity 36, 157–159 (2012).
Linossi, E. M. et al. Discovery of an exosite on the SOCS2-SH2 domain that enhances SH2 binding to phosphorylated ligands. Nat. Commun. 12, 7032 (2021).
Durham, G. A., Williams, J. J. L., Nasim, M. T. & Palmer, T. M. Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharm. Sci. 40, 298–308 (2019).
Liau, N. P. D. et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 9, 1558 (2018).
Rico-Bautista, E., Flores-Morales, A. & Fernández-Pérez, L. Suppressor of cytokine signaling (SOCS) 2, a protein with multiple functions. Cytokine Growth Factor Rev. 17, 431–439 (2006).
Yoshimura, A., Naka, T. & Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 7, 454–465 (2007).
Starr, R. et al. A family of cytokine-inducible inhibitors of signalling. Nature 387, 917–921 (1997).
Liang, Y., Xu, W. D., Peng, H., Pan, H. F. & Ye, D. Q. SOCS signaling in autoimmune diseases: molecular mechanisms and therapeutic implications. Eur. J. Immunol. 44, 1265–1275 (2014).
Alexander, W. S. et al. Suppressors of cytokine signaling (SOCS): negative regulators of signal transduction. J. Leukoc. Biol. 66, 588–592 (1999).
Kishimoto, T. & Kikutani, H. Knocking the SOCS off a tumor suppressor. Nat. Genet. 28, 4–5 (2001).
Thomas, S. J., Snowden, J. A., Zeidler, M. P. & Danson, S. J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 113, 365–371 (2015).
Seif, F. et al. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 15, 23 (2017).
Wu, C. S. & Zou, L. The SUMO (small ubiquitin-like modifier) ligase PIAS3 primes ATR for checkpoint activation. J. Biol. Chem. 291, 279–290 (2016).
Rytinki, M. M., Kaikkonen, S., Pehkonen, P., Jääskeläinen, T. & Palvimo, J. J. PIAS proteins: pleiotropic interactors associated with SUMO. Cell Mol. Life Sci. 66, 3029–3041 (2009).
Frankson, R. et al. Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res. 77, 5701–5705 (2017).
Pike, K. A. & Tremblay, M. L. Protein tyrosine phosphatases: regulators of CD4 T cells in inflammatory bowel disease. Front. Immunol. 9, 2504 (2018).
Long, D., Chen, Y., Wu, H., Zhao, M. & Lu, Q. Clinical significance and immunobiology of IL-21 in autoimmunity. J. Autoimmun. 99, 1–14 (2019).
Yan, Z., Gibson, S. A., Buckley, J. A., Qin, H. & Benveniste, E. N. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin. Immunol. 189, 4–13 (2018).
Roxburgh, C. S. & McMillan, D. C. Therapeutics targeting innate immune/inflammatory responses through the interleukin-6/JAK/STAT signal transduction pathway in patients with cancer. Transl. Res. 167, 61–66 (2016).
O’Shea, J. J. & Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36, 542–550 (2012).
O’Shea, J. J. & Murray, P. J. Cytokine signaling modules in inflammatory responses. Immunity 28, 477–487 (2008).
Darnell, J. E. Jr., Kerr, I. M. & Stark, G. R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421 (1994).
Schwartz, D. M., Bonelli, M., Gadina, M. & O’Shea, J. J. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat. Rev. Rheumatol. 12, 25–36 (2016).
O’Shea, J. J., Holland, S. M. & Staudt, L. M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 368, 161–170 (2013).
Costa-Pereira, A. P. et al. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc. Natl Acad. Sci. USA 99, 8043–8047 (2002).
O’Shea, J. J., Kontzias, A., Yamaoka, K., Tanaka, Y. & Laurence, A. Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 72, ii111–ii115 (2013).
Kang, Y. H., Biswas, A., Field, M. & Snapper, S. B. STAT1 signaling shields T cells from NK cell-mediated cytotoxicity. Nat. Commun. 10, 912 (2019).
Putz, E. M. et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep. 4, 437–444 (2013).
Watford, W. T. et al. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol. Rev. 202, 139–156 (2004).
Boudreau, J. E. et al. IL-15 and type I interferon are required for activation of tumoricidal NK cells by virus-infected dendritic cells. Cancer Res. 71, 2497–2506 (2011).
Cordell, H. J. et al. An international genome-wide meta-analysis of primary biliary cholangitis: novel risk loci and candidate drugs. J. Hepatol. 75, 572–581 (2021).
Goropevšek, A., Holcar, M. & Avčin, T. The role of STAT signaling pathways in the pathogenesis of systemic lupus erythematosus. Clin. Rev. Allergy Immunol. 52, 164–181 (2017).
Park, J. S. et al. STA-21, a promising STAT-3 inhibitor that reciprocally regulates Th17 and Treg cells, inhibits osteoclastogenesis in mice and humans and alleviates autoimmune inflammation in an experimental model of rheumatoid arthritis. Arthritis Rheumatol. 66, 918–929 (2014).
Ma, C. S. et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood 119, 3997–4008 (2012).
Gracey, E. et al. TYK2 inhibition reduces type 3 immunity and modifies disease progression in murine spondyloarthritis. J. Clin. Invest. 130, 1863–1878 (2020).
Sigurdsson, S. et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am. J. Hum. Genet. 76, 528–537 (2005).
Ellinghaus, D. et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am. J. Hum. Genet. 90, 636–647 (2012).
Casaca, V. I. et al. STAT6 polymorphisms are associated with neonatal regulatory T cells and cytokines and atopic diseases at 3 years. Allergy 68, 1249–1258 (2013).
Fabre, A. et al. Clinical aspects of STAT3 gain-of-function germline mutations: a systematic review. J. Allergy Clin. Immunol. Pr. 7, 1958–1969.e1959 (2019).
Lundgren, S. et al. Somatic mutations in lymphocytes in patients with immune-mediated aplastic anemia. Leukemia 35, 1365–1379 (2021).
Marabelle, A., Aspeslagh, S., Postel-Vinay, S. & Soria, J. C. JAK mutations as escape mechanisms to anti-PD-1 therapy. Cancer Disco. 7, 128–130 (2017).
Jehanno, C., Vulin, M., Richina, V., Richina, F. & Bentires-Alj, M. Phenotypic plasticity during metastatic colonization. Trends Cell Biol. 32, 854–867 (2022).
Westneat, D. F., Potts, L. J., Sasser, K. L. & Shaffer, J. D. Causes and consequences of phenotypic plasticity in complex environments. Trends Ecol. Evol. 34, 555–568 (2019).
Bakir, B., Chiarella, A. M., Pitarresi, J. R. & Rustgi, A. K. EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol. 30, 764–776 (2020).
Burkhardt, D. B. et al. Mapping phenotypic plasticity upon the cancer cell state landscape using manifold learning. Cancer Discov. 12, 1847–1859 (2022).
Lee, Y. I. et al. WNT5A drives interleukin-6-dependent epithelial-mesenchymal transition via the JAK/STAT pathway in keloid pathogenesis. Burns Trauma 10, tkac023 (2022).
Quintanal-Villalonga, Á. et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 17, 360–371 (2020).
Yu, H. A. et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 19, 2240–2247 (2013).
Beebe, K., Lee, W. C. & Micchelli, C. A. JAK/STAT signaling coordinates stem cell proliferation and multilineage differentiation in the Drosophila intestinal stem cell lineage. Dev. Biol. 338, 28–37 (2010).
Lo, U. G. et al. The driver role of JAK-STAT signalling in cancer stemness capabilities leading to new therapeutic strategies for therapy- and castration-resistant prostate cancer. Clin. Transl. Med. 12, e978 (2022).
Chatterjee, D. et al. Cell polarity opposes Jak/STAT-mediated Escargot activation that drives intratumor heterogeneity in a Drosophila tumor model. Cell Rep. 42, 112061 (2023).
Deng, S. et al. Ectopic JAK-STAT activation enables the transition to a stem-like and multilineage state conferring AR-targeted therapy resistance. Nat. Cancer 3, 1071–1087 (2022).
Sun, L. et al. Modelling liver cancer initiation with organoids derived from directly reprogrammed human hepatocytes. Nat. Cell Biol. 21, 1015–1026 (2019).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Baratchian, M. et al. H3K9 methylation drives resistance to androgen receptor-antagonist therapy in prostate cancer. Proc. Natl Acad. Sci. USA 119, e2114324119 (2022).
Firestein, G. S. Evolving concepts of rheumatoid arthritis. Nature 423, 356–361 (2003).
Scott, D. L., Wolfe, F. & Huizinga, T. W. Rheumatoid arthritis. Lancet 376, 1094–1108 (2010).
Scherer, H. U., Häupl, T. & Burmester, G. R. The etiology of rheumatoid arthritis. J. Autoimmun. 110, 102400 (2020).
McInnes, I. B. & Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 365, 2205–2219 (2011).
Noack, M. & Miossec, P. Selected cytokine pathways in rheumatoid arthritis. Semin. Immunopathol. 39, 365–383 (2017).
Pieringer, H. & Studnicka-Benke, A. What is causing my arthritis, doctor? A glimpse beyond the usual suspects in the pathogenesis of rheumatoid arthritis. QJM 106, 219–228 (2013).
Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 11, 415–429 (2015).
Genovese, M. C. et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease-modifying antirheumatic drug therapy: the FINCH 2 randomized clinical trial. Jama 322, 315–325 (2019).
Kavanaugh, A. et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann. Rheum. Dis. 76, 1009–1019 (2017).
Combe, B. et al. Filgotinib versus placebo or adalimumab in patients with rheumatoid arthritis and inadequate response to methotrexate: a phase III randomised clinical trial. Ann. Rheum. Dis. 80, 848–858 (2021).
Nguyen, H. N. et al. Autocrine loop involving IL-6 family member LIF, LIF Receptor, and STAT4 drives sustained fibroblast production of inflammatory mediators. Immunity 46, 220–232 (2017).
Mori, T. et al. IL-1β and TNFα-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int. Immunol. 23, 701–712 (2011).
Kasperkovitz, P. V. et al. Activation of the STAT1 pathway in rheumatoid arthritis. Ann. Rheum. Dis. 63, 233–239 (2004).
van der Pouw Kraan, T. C. et al. Rheumatoid arthritis is a heterogeneous disease: evidence for differences in the activation of the STAT-1 pathway between rheumatoid tissues. Arthritis Rheum. 48, 2132–2145 (2003).
Walker, J. G. et al. Expression of Jak3, STAT1, STAT4, and STAT6 in inflammatory arthritis: unique Jak3 and STAT4 expression in dendritic cells in seropositive rheumatoid arthritis. Ann. Rheum. Dis. 65, 149–156 (2006).
Tanaka, Y. et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to conventional DMARDs: a randomised, double-blind, placebo-controlled phase III trial (RAJ3). Ann. Rheum. Dis. 78, 1320–1332 (2019).
Takeuchi, T. et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III randomised, double-blind, placebo-controlled trial (RAJ4) in Japan. Ann. Rheum. Dis. 78, 1305–1319 (2019).
Takeuchi, T. et al. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann. Rheum. Dis. 75, 1057–1064 (2016).
Genovese, M. C. et al. Peficitinib, a JAK inhibitor, in combination with limited conventional synthetic disease-modifying antirheumatic drugs in the treatment of moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol. 69, 932–942 (2017).
van der Pouw Kraan, T. C. et al. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann. Rheum. Dis. 66, 1008–1014 (2007).
Higgs, B. W. et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann. Rheum. Dis. 70, 2029–2036 (2011).
Wang, J. & Zhao, Q. LncRNA LINC-PINT increases SOCS1 expression by sponging miR-155-5p to inhibit the activation of ERK signaling pathway in rheumatoid arthritis synovial fibroblasts induced by TNF-α. Int. Immunopharmacol. 84, 106497 (2020).
Kivitz, A. J. et al. Peficitinib, a JAK inhibitor, in the treatment of moderate-to-severe rheumatoid arthritis in patients with an inadequate response to methotrexate. Arthritis Rheumatol. 69, 709–719 (2017).
Papp, K. et al. A phase 2a randomized, double-blind, placebo-controlled, sequential dose-escalation study to evaluate the efficacy and safety of ASP015K, a novel Janus kinase inhibitor, in patients with moderate-to-severe psoriasis. Br. J. Dermatol. 173, 767–776 (2015).
Sands, B. E. et al. Peficitinib, an oral janus kinase inhibitor, in moderate-to-severe ulcerative colitis: results from a randomised, phase 2 study. J. Crohns Colitis 12, 1158–1169 (2018).
Lauper, K. et al. Effectiveness of TNF-inhibitors, abatacept, IL6-inhibitors and JAK-inhibitors in 31 846 patients with rheumatoid arthritis in 19 registers from the ‘JAK-pot’ collaboration. Ann. Rheum. Dis. 81, 1358–1366 (2022).
Westhovens, R. et al. Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose-finding study (DARWIN 1). Ann. Rheum. Dis. 76, 998–1008 (2017).
Vanhoutte, F. et al. Efficacy, safety, pharmacokinetics, and pharmacodynamics of filgotinib, a selective JAK-1 inhibitor, after short-term treatment of rheumatoid arthritis: results of two randomized phase IIa trials. Arthritis Rheumatol. 69, 1949–1959 (2017).
Westhovens, R. et al. Filgotinib in combination with methotrexate or as monotherapy versus methotrexate monotherapy in patients with active rheumatoid arthritis and limited or no prior exposure to methotrexate: the phase 3, randomised controlled FINCH 3 trial. Ann. Rheum. Dis. 80, 727–738 (2021).
Chen, C. et al. A highly selective JAK3 inhibitor is developed for treating rheumatoid arthritis by suppressing γc cytokine-related JAK-STAT signal. Sci. Adv. 8, eabo4363 (2022).
Charles-Schoeman, C. et al. Efficacy and safety of tofacitinib following inadequate response to conventional synthetic or biological disease-modifying antirheumatic drugs. Ann. Rheum. Dis. 75, 1293–1301 (2016).
Fleischmann, R. et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 64, 617–629 (2012).
van Vollenhoven, R. F. et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N. Engl. J. Med. 367, 508–519 (2012).
Kremer, J. et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann. Intern. Med. 159, 253–261 (2013).
van der Heijde, D. et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 65, 559–570 (2013).
Lee, E. B. et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N. Engl. J. Med. 370, 2377–2386 (2014).
Mesa, R. A. et al. SIMPLIFY-1: a phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor-naïve patients with myelofibrosis. J. Clin. Oncol. 35, 3844–3850 (2017).
Mesa, R. et al. Overall survival in the SIMPLIFY-1 and SIMPLIFY-2 phase 3 trials of momelotinib in patients with myelofibrosis. Leukemia 36, 2261–2268 (2022).
Meyer, S. C. et al. CHZ868, a type II JAK2 inhibitor, reverses type I JAK inhibitor persistence and demonstrates efficacy in myeloproliferative neoplasms. Cancer Cell 28, 15–28 (2015).
Harrison, C. N. et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 5, e73–e81 (2018).
Tefferi, A. & Vardiman, J. W. Myelodysplastic syndromes. N. Engl. J. Med. 361, 1872–1885 (2009).
Zeidan, A. M., Shallis, R. M., Wang, R., Davidoff, A. & Ma, X. Epidemiology of myelodysplastic syndromes: why characterizing the beast is a prerequisite to taming it. Blood Rev. 34, 1–15 (2019).
Aul, C., Bowen, D. T. & Yoshida, Y. Pathogenesis, etiology and epidemiology of myelodysplastic syndromes. Haematologica 83, 71–86 (1998).
Cazzola, M. Myelodysplastic syndromes. N. Engl. J. Med 383, 1358–1374 (2020).
Bejar, R. et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 364, 2496–2506 (2011).
Tefferi, A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 24, 1128–1138 (2010).
Quentmeier, H. et al. SOCS2: inhibitor of JAK2V617F-mediated signal transduction. Leukemia 22, 2169–2175 (2008).
Plo, I. et al. JAK2 stimulates homologous recombination and genetic instability: potential implication in the heterogeneity of myeloproliferative disorders. Blood 112, 1402–1412 (2008).
Dawson, M. A. et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 461, 819–822 (2009).
Thiele, J. et al. The international consensus classification of myeloid neoplasms and acute Leukemias: myeloproliferative neoplasms. Am. J. Hematol. 98, 166–179 (2023).
Chifotides, H. T., Bose, P. & Verstovsek, S. Momelotinib: an emerging treatment for myelofibrosis patients with anemia. J. Hematol. Oncol. 15, 7 (2022).
Tefferi, A. et al. Momelotinib for myelofibrosis: 12-year survival data and retrospective comparison to ruxolitinib. Am. J. Hematol. 97, E433–e435 (2022).
Geyer, H. L. & Mesa, R. A. Therapy for myeloproliferative neoplasms: when, which agent, and how? Blood 124, 3529–3537 (2014).
Rampal, R. et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 123, e123–e133 (2014).
Jones, A. V. et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 106, 2162–2168 (2005).
Ungureanu, D. et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat. Struct. Mol. Biol. 18, 971–976 (2011).
Kralovics, R. et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790 (2005).
Woods, B. et al. Activation of JAK/STAT signaling in megakaryocytes sustains myeloproliferation in vivo. Clin. Cancer Res. 25, 5901–5912 (2019).
Arber, D. A. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405 (2016).
Kollmann, K. et al. A novel signalling screen demonstrates that CALR mutations activate essential MAPK signalling and facilitate megakaryocyte differentiation. Leukemia 31, 934–944 (2017).
Tefferi, A. et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia 28, 1472–1477 (2014).
Pikman, Y. et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 3, e270 (2006).
Nangalia, J. et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 369, 2391–2405 (2013).
Klampfl, T. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 369, 2379–2390 (2013).
Elf, S. et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 6, 368–381 (2016).
Mullally, A., Lane, S. W., Brumme, K. & Ebert, B. L. Myeloproliferative neoplasm animal models. Hematol. Oncol. Clin. North Am. 26, 1065–1081 (2012).
Debili, N. et al. The Mpl-ligand or thrombopoietin or megakaryocyte growth and differentiative factor has both direct proliferative and differentiative activities on human megakaryocyte progenitors. Blood 86, 2516–2525 (1995).
Kaushansky, K. et al. Thrombopoietin, the Mp1 ligand, is essential for full megakaryocyte development. Proc. Natl Acad. Sci. USA 92, 3234–3238 (1995).
Ku, H., Yonemura, Y., Kaushansky, K. & Ogawa, M. Thrombopoietin, the ligand for the Mpl receptor, synergizes with steel factor and other early acting cytokines in supporting proliferation of primitive hematopoietic progenitors of mice. Blood 87, 4544–4551 (1996).
Lussana, F. et al. Driver mutations (JAK2V617F, MPLW515L/K or CALR), pentraxin-3 and C-reactive protein in essential thrombocythemia and polycythemia vera. J. Hematol. Oncol. 10, 54 (2017).
Lasho, T. L. et al. Concurrent MPL515 and JAK2V617F mutations in myelofibrosis: chronology of clonal emergence and changes in mutant allele burden over time. Br. J. Haematol. 135, 683–687 (2006).
Pardanani, A. D. et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 108, 3472–3476 (2006).
Langan, S. M., Irvine, A. D. & Weidinger, S. Atopic dermatitis. Lancet 396, 345–360 (2020).
Eckert, L. et al. The burden of atopic dermatitis in US adults: health care resource utilization data from the 2013 National Health and Wellness Survey. J. Am. Acad. Dermatol. 78, 54–61.e51 (2018).
Drucker, A. M., Wang, A. R. & Qureshi, A. A. Research gaps in quality of life and economic burden of atopic dermatitis: the national eczema association burden of disease audit. JAMA Dermatol. 152, 873–874 (2016).
Eichenfield, L. F. et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J. Am. Acad. Dermatol. 70, 338–351 (2014).
Freitas, E., Gooderham, M. & Torres, T. New topical therapies in development for atopic dermatitis. Drugs 82, 843–853 (2022).
Tsoi, L. C. et al. Progression of acute-to-chronic atopic dermatitis is associated with quantitative rather than qualitative changes in cytokine responses. J. Allergy Clin. Immunol. 145, 1406–1415 (2020).
Huang, I. H., Chung, W. H., Wu, P. C. & Chen, C. B. JAK-STAT signaling pathway in the pathogenesis of atopic dermatitis: an updated review. Front. Immunol. 13, 1068260 (2022).
Makowska, K. et al. Immunopathogenesis of atopic dermatitis: focus on interleukins as disease drivers and therapeutic targets for novel treatments. Int. J. Mol. Sci. 24, 781 (2023).
Cotter, D. G., Schairer, D. & Eichenfield, L. Emerging therapies for atopic dermatitis: JAK inhibitors. J. Am. Acad. Dermatol. 78, S53–s62 (2018).
Nakashima, C., Yanagihara, S. & Otsuka, A. Innovation in the treatment of atopic dermatitis: emerging topical and oral Janus kinase inhibitors. Allergol. Int. 71, 40–46 (2022).
Kopalli, S. R., Annamneedi, V. P. & Koppula, S. Potential natural biomolecules targeting JAK/STAT/SOCS signaling in the management of atopic dermatitis. Molecules 27, 4660 (2022).
Klaeschen, A. S. et al. JAK1/2 inhibition impairs the development and function of inflammatory dendritic epidermal cells in atopic dermatitis. J. Allergy Clin. Immunol. 147, 2202–2212.e2208 (2021).
Reich, K. et al. Efficacy and safety of baricitinib combined with topical corticosteroids for treatment of moderate to severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 156, 1333–1343 (2020).
Simpson, E. L. et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br. J. Dermatol. 183, 242–255 (2020).
Papp, K. et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: results from 2 phase 3, randomized, double-blind studies. J. Am. Acad. Dermatol. 85, 863–872 (2021).
Papp, K. et al. Long-term safety and disease control with ruxolitinib cream in atopic dermatitis: results from two phase 3 studies. J. Am. Acad. Dermatol. 88, 1008–1016 (2022).
Kim, B. S. et al. Treatment of atopic dermatitis with ruxolitinib cream (JAK1/JAK2 inhibitor) or triamcinolone cream. J. Allergy Clin. Immunol. 145, 572–582 (2020).
Reich, K. et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 397, 2169–2181 (2021).
Rubbert-Roth, A. et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N. Engl. J. Med. 383, 1511–1521 (2020).
Cohen, S. B. et al. Safety profile of upadacitinib in rheumatoid arthritis: integrated analysis from the SELECT phase III clinical programme. Ann. Rheum. Dis. 80, 304–311 (2021).
Fleischmann, R. M. et al. Safety and effectiveness of upadacitinib or adalimumab plus methotrexate in patients with rheumatoid arthritis over 48 weeks with switch to alternate therapy in patients with insufficient response. Ann. Rheum. Dis. 78, 1454–1462 (2019).
Mendes-Bastos, P. et al. Characterization of acne associated with upadacitinib treatment in patients with moderate-to-severe atopic dermatitis: A post hoc integrated analysis of 3 phase 3 randomized, double-blind, placebo-controlled trials. J. Am. Acad. Dermatol. 87, 784–791 (2022).
Deodhar, A. et al. Safety and efficacy of upadacitinib in patients with active ankylosing spondylitis and an inadequate response to nonsteroidal antiinflammatory drug therapy: one-year results of a double-blind, placebo-controlled study and open-label extension. Arthritis Rheumatol. 74, 70–80 (2022).
van Vollenhoven, R. et al. Efficacy and safety of upadacitinib monotherapy in methotrexate-naive patients with moderately-to-severely active rheumatoid arthritis (SELECT-EARLY): a multicenter, multi-country, randomized, double-blind, active comparator-controlled trial. Arthritis Rheumatol. 72, 1607–1620 (2020).
Fleischmann, R. et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase iii, double-blind, randomized controlled trial. Arthritis Rheumatol. 71, 1788–1800 (2019).
Silverberg, J. I. et al. Upadacitinib plus topical corticosteroids in atopic dermatitis: week 52 AD Up study results. J. Allergy Clin. Immunol. 149, 977–987.e914 (2022).
Guttman-Yassky, E. et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 145, 877–884 (2020).
Forbes, G. M. In active UC, upadacitinib induced and maintained remission. Ann. Intern. Med. 175, Jc113 (2022).
Sandborn, W. J. et al. Efficacy of upadacitinib in a randomized trial of patients with active ulcerative colitis. Gastroenterology 158, 2139–2149.e2114 (2020).
Sandborn, W. J. et al. Efficacy and safety of upadacitinib in a randomized trial of patients with Crohn’s disease. Gastroenterology 158, 2123–2138.e2128 (2020).
Mease, P. J. et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann. Rheum. Dis. 80, 312–320 (2021).
He, H. & Guttman-Yassky, E. JAK inhibitors for atopic dermatitis: an update. Am. J. Clin. Dermatol. 20, 181–192 (2019).
Mohamed, M. F. et al. Pharmacokinetics, safety and tolerability of ABT-494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin. Pharmacokinet. 55, 1547–1558 (2016).
van der Heijde, D. et al. Efficacy and safety of upadacitinib for active ankylosing spondylitis refractory to biological therapy: a double-blind, randomised, placebo-controlled phase 3 trial. Ann. Rheum. Dis. 81, 1515–1523 (2022).
Fleischmann, R. M. et al. Switching between Janus kinase inhibitor upadacitinib and adalimumab following insufficient response: efficacy and safety in patients with rheumatoid arthritis. Ann. Rheum. Dis. 80, 432–439 (2021).
McInnes, I. B. et al. Trial of upadacitinib and adalimumab for psoriatic arthritis. N. Engl. J. Med 384, 1227–1239 (2021).
Ghosh, S. et al. Upadacitinib treatment improves symptoms of bowel urgency and abdominal pain, and correlates with quality of life improvements in patients with moderate to severe ulcerative colitis. J. Crohns Colitis 15, 2022–2030 (2021).
Guttman-Yassky, E. et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet 397, 2151–2168 (2021).
Vazquez, M. L. et al. Identification of N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfonamide (PF-04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J. Med. Chem. 61, 1130–1152 (2018).
Mikhaylov, D., Ungar, B., Renert-Yuval, Y. & Guttman-Yassky, E. Oral Janus kinase inhibitors for atopic dermatitis. Ann. Allergy Asthma Immunol. 130, 577–592 (2023).
De, S. K. Abrocitinib: first globally approved selective janus kinase-1 inhibitor for the treatment of atopic dermatitis. Curr. Med. Chem. 30, https://doi.org/10.2174/0929867330666230216123419, E-pub Ahead of Print (2023).
Deeks, E. D. & Duggan, S. Abrocitinib: first approval. Drugs 81, 2149–2157 (2021).
Bieber, T. et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N. Engl. J. Med. 384, 1101–1112 (2021).
Blauvelt, A. et al. Abrocitinib induction, randomized withdrawal, and retreatment in patients with moderate-to-severe atopic dermatitis: results from the JAK1 atopic dermatitis efficacy and safety (JADE) REGIMEN phase 3 trial. J. Am. Acad. Dermatol. 86, 104–112 (2022).
Weidinger, S. & Schreiber, S. Abrocitinib for atopic dermatitis: a step forward. Lancet 396, 215–217 (2020).
Simpson, E. L. et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet 396, 255–266 (2020).
Uppal, S. K., Chat, V. S., Kearns, D. G. & Wu, J. J. Abrocitinib for atopic dermatitis. Lancet 397, 195–196 (2021).
Schmieder, G. J. et al. Efficacy and safety of the Janus kinase 1 inhibitor PF-04965842 in patients with moderate-to-severe psoriasis: phase II, randomized, double-blind, placebo-controlled study. Br. J. Dermatol. 179, 54–62 (2018).
Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 380, 1450–1462 (2019).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Altekruse, S. F., McGlynn, K. A. & Reichman, M. E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. 27, 1485–1491 (2009).
Llovet, J. M. et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 7, 6 (2021).
European Association For The Study Of The Liver & European Organisation For Research And Treatment Of Cancer. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J. Hepatol. 56, 908–943 (2012).
Bruix, J. & Sherman, M. Management of hepatocellular carcinoma. Hepatology 42, 1208–1236 (2005).
Schafer, D. F. & Sorrell, M. F. Hepatocellular carcinoma. Lancet 353, 1253–1257 (1999).
Parikh, N. D. & Pillai, A. Recent advances in hepatocellular carcinoma treatment. Clin. Gastroenterol. Hepatol. 19, 2020–2024 (2021).
Caraglia, M. et al. Alpha-interferon and its effects on signalling pathways within cells. Curr. Protein Pept. Sci. 5, 475–485 (2004).
Wang, Y. X. et al. Interferon-inducible MX2 is a host restriction factor of hepatitis B virus replication. J. Hepatol. 72, 865–876 (2020).
Han, M. et al. Altered expression of interferon-stimulated genes is strongly associated with therapeutic outcomes in hepatitis B virus infection. Antivir. Res. 147, 75–85 (2017).
Koeberlein, B. et al. Hepatitis B virus overexpresses suppressor of cytokine signaling-3 (SOCS3) thereby contributing to severity of inflammation in the liver. Virus Res. 148, 51–59 (2010).
Robek, M. D., Boyd, B. S., Wieland, S. F. & Chisari, F. V. Signal transduction pathways that inhibit hepatitis B virus replication. Proc. Natl Acad. Sci. USA 101, 1743–1747 (2004).
Gao, D. et al. Down-regulation of suppressor of cytokine signaling 3 by miR-122 enhances interferon-mediated suppression of hepatitis B virus. Antivir. Res. 118, 20–28 (2015).
Khan, M. G. M. et al. Prognostic significance of SOCS1 and SOCS3 tumor suppressors and oncogenic signaling pathway genes in hepatocellular carcinoma. BMC Cancer 20, 774 (2020).
Li, H. et al. C1QTNF1-AS1 regulates the occurrence and development of hepatocellular carcinoma by regulating miR-221-3p/SOCS3. Hepatol. Int. 13, 277–292 (2019).
Liu, Z. K. et al. EYA2 suppresses the progression of hepatocellular carcinoma via SOCS3-mediated blockade of JAK/STAT signaling. Mol. Cancer 20, 79 (2021).
Zhao, Z. et al. CircSOD2 induced epigenetic alteration drives hepatocellular carcinoma progression through activating JAK2/STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 39, 259 (2020).
Guo, H. et al. HHLA2 activates the JAK/STAT signaling pathway by binding to TMIGD2 in hepatocellular carcinoma cells. Inflammation 45, 1585–1599 (2022).
Mora, L. B. et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 62, 6659–6666 (2002).
Gao, L. et al. Down-regulation of signal transducer and activator of transcription 3 expression using vector-based small interfering RNAs suppresses growth of human prostate tumor in vivo. Clin. Cancer Res. 11, 6333–6341 (2005).
Liu, C. et al. Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. Prostate 75, 1341–1353 (2015).
Liu, C. et al. Inhibition of constitutively active Stat3 reverses enzalutamide resistance in LNCaP derivative prostate cancer cells. Prostate 74, 201–209 (2014).
Onkar, S. S. et al. The Great Immune Escape: Understanding the Divergent Immune Response in Breast Cancer Subtypes. Cancer Discov. 13, 23–40 (2023).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 (2019).
Feng, R. et al. Cancer situation in China: what does the China cancer map indicate from the first national death survey to the latest cancer registration? Cancer Commun. (Lond.) 43, 75–86 (2023).
Luen, S. J. et al. Genomic characterisation of hormone receptor-positive breast cancer arising in very young women. Ann Oncol, (2023).
Chlebowski, R. T. et al. Ethnicity and breast cancer: factors influencing differences in incidence and outcome. J. Natl Cancer Inst. 97, 439–448 (2005).
Wong, G. L., Manore, S. G., Doheny, D. L. & Lo, H. W. STAT family of transcription factors in breast cancer: Pathogenesis and therapeutic opportunities and challenges. Semin. Cancer Biol. 86, 84–106 (2022).
Zou, S. et al. Targeting STAT3 in cancer immunotherapy. Mol. Cancer 19, 145 (2020).
Bromberg, J. Signal transducers and activators of transcription as regulators of growth, apoptosis and breast development. Breast Cancer Res. 2, 86–90 (2000).
Yu, H., Lee, H., Herrmann, A., Buettner, R. & Jove, R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer 14, 736–746 (2014).
Johnson, D. E., O’Keefe, R. A. & Grandis, J. R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 15, 234–248 (2018).
Wang, T. et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 27, 136–150.e135 (2018).
Li, J. et al. Tumor cell-intrinsic CD96 mediates chemoresistance and cancer stemness by regulating mitochondrial fatty acid β-oxidation. Adv. Sci. (Weinh.) 10, e2202956 (2023).
Zhu, Y. et al. Natural product preferentially targets redox and metabolic adaptations and aberrantly active STAT3 to inhibit breast tumor growth in vivo. Cell Death Dis. 13, 1022 (2022).
Liu, C. et al. A highly potent small-molecule antagonist of exportin-1 selectively eliminates CD44(+)CD24(-) enriched breast cancer stem-like cells. Drug Resist. Updat. 66, 100903 (2023).
Meraviglia-Crivelli, D. et al. IL-6/STAT3 signaling in tumor cells restricts the expression of frameshift-derived neoantigens by SMG1 induction. Mol. Cancer 21, 211 (2022).
Dinakar, Y. H. et al. Role of STAT3 in the initiation, progression, proliferation and metastasis of breast cancer and strategies to deliver JAK and STAT3 inhibitors. Life Sci. 309, 120996 (2022).
DeMichele, A. et al. Host genetic variants in the interleukin-6 promoter predict poor outcome in patients with estrogen receptor-positive, node-positive breast cancer. Cancer Res. 69, 4184–4191 (2009).
Dethlefsen, C., Højfeldt, G. & Hojman, P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res. Treat. 138, 657–664 (2013).
Rodriguez-Barrueco, R. et al. Inhibition of the autocrine IL-6-JAK2-STAT3-calprotectin axis as targeted therapy for HR-/HER2+ breast cancers. Genes Dev. 29, 1631–1648 (2015).
Bottos, A. et al. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat. Commun. 7, 12258 (2016).
Irey, E. A. et al. JAK/STAT inhibition in macrophages promotes therapeutic resistance by inducing expression of protumorigenic factors. Proc. Natl Acad. Sci. USA 116, 12442–12451 (2019).
Tao, J. et al. JAK-STAT signaling is activated in the kidney and peripheral blood cells of patients with focal segmental glomerulosclerosis. Kidney Int. 94, 795–808 (2018).
Pang, M. et al. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int. 78, 257–268 (2010).
Koike, K. et al. Protective role of JAK/STAT signaling against renal fibrosis in mice with unilateral ureteral obstruction. Clin. Immunol. 150, 78–87 (2014).
Luan, J. et al. miR-150-based RNA interference attenuates tubulointerstitial fibrosis through the SOCS1/JAK/STAT pathway in vivo and in vitro. Mol. Ther. Nucleic Acids 22, 871–884 (2020).
Tao, J. et al. JAK-STAT activity in peripheral blood cells and kidney tissue in IgA nephropathy. Clin. J. Am. Soc. Nephrol. 15, 973–982 (2020).
Wang, X., Shaw, S., Amiri, F., Eaton, D. C. & Marrero, M. B. Inhibition of the Jak/STAT signaling pathway prevents the high glucose-induced increase in tgf-beta and fibronectin synthesis in mesangial cells. Diabetes 51, 3505–3509 (2002).
Marrero, M. B., Banes-Berceli, A. K., Stern, D. M. & Eaton, D. C. Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 290, F762–F768 (2006).
Sengupta, U., Ukil, S., Dimitrova, N. & Agrawal, S. Expression-based network biology identifies alteration in key regulatory pathways of type 2 diabetes and associated risk/complications. PLoS One 4, e8100 (2009).
Chen, D. et al. JAK/STAT pathway promotes the progression of diabetic kidney disease via autophagy in podocytes. Eur. J. Pharm. 902, 174121 (2021).
Zhang, H. et al. Podocyte-specific JAK2 overexpression worsens diabetic kidney disease in mice. Kidney Int. 92, 909–921 (2017).
Casarella, A. et al. Autosomic dominant polycystic kidney disease and metformin: old knowledge and new insights on retarding progression of chronic kidney disease. Med. Res. Rev. 42, 629–640 (2022).
Fragiadaki, M. et al. STAT5 drives abnormal proliferation in autosomal dominant polycystic kidney disease. Kidney Int 91, 575–586 (2017).
Patera, F., Cudzich-Madry, A., Huang, Z. & Fragiadaki, M. Renal expression of JAK2 is high in polycystic kidney disease and its inhibition reduces cystogenesis. Sci. Rep. 9, 4491 (2019).
Arakawa, T. et al. Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrol. Dial. Transpl. 23, 3418–3426 (2008).
Borie, D. C. et al. Combined use of the JAK3 inhibitor CP-690,550 with mycophenolate mofetil to prevent kidney allograft rejection in nonhuman primates. Transplantation 80, 1756–1764 (2005).
Borie, D. C. et al. Immunosuppression by the JAK3 inhibitor CP-690,550 delays rejection and significantly prolongs kidney allograft survival in nonhuman primates. Transplantation 79, 791–801 (2005).
Changelian, P. S. et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 302, 875–878 (2003).
Adachi, Y. et al. Inhibition of FGFR reactivates IFNγ signaling in tumor cells to enhance the combined antitumor activity of lenvatinib with anti-PD-1 antibodies. Cancer Res. 82, 292–306 (2022).
Favoino, E. et al. Working and safety profiles of JAK/STAT signaling inhibitors. Are these small molecules also smart? Autoimmun. Rev. 20, 102750 (2021).
Podewski, E. K. et al. Alterations in Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling in patients with end-stage dilated cardiomyopathy. Circulation 107, 798–802 (2003).
Panés, J. & Vermeire, S. JAK inhibitors: back to small molecules for the treatment of IBD. J. Crohns Colitis 14, S711–s712 (2020).
Miyagi, T. et al. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 204, 2383–2396 (2007).
Thieu, V. T. et al. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity 29, 679–690 (2008).
Weinstein, J. S. et al. STAT4 and T-bet control follicular helper T cell development in viral infections. J. Exp. Med. 215, 337–355 (2018).
Guenterberg, K. D. et al. Interleukin-29 binds to melanoma cells inducing Jak-STAT signal transduction and apoptosis. Mol. Cancer Ther. 9, 510–520 (2010).
Kroon, P. et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 73, 5288–5298 (2013).
Tanaka, T., Narazaki, M., Ogata, A. & Kishimoto, T. A new era for the treatment of inflammatory autoimmune diseases by interleukin-6 blockade strategy. Semin. Immunol. 26, 88–96 (2014).
Chen, L. Y. C. et al. Soluble interleukin-6 receptor in the COVID-19 cytokine storm syndrome. Cell Rep. Med. 2, 100269 (2021).
Refaeli, Y., Van Parijs, L., London, C. A., Tschopp, J. & Abbas, A. K. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity 8, 615–623 (1998).
Liu, X. et al. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 11, 179–186 (1997).
Lin, J. X. & Leonard, W. J. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 19, 2566–2576 (2000).
Mui, A. L., Wakao, H., O’Farrell, A. M., Harada, N. & Miyajima, A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J. 14, 1166–1175 (1995).
Wakao, H., Gouilleux, F. & Groner, B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. 14, 854–855 (1995).
Azam, M. et al. Interleukin-3 signals through multiple isoforms of Stat5. EMBO J. 14, 1402–1411 (1995).
Buettner, R., Mora, L. B. & Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 8, 945–954 (2002).
Page, B. D. et al. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J. Med Chem. 55, 1047–1055 (2012).
Lis, C. et al. Development of Erasin: a chromone-based STAT3 inhibitor which induces apoptosis in Erlotinib-resistant lung cancer cells. Sci. Rep. 7, 17390 (2017).
Caldenhoven, E. et al. STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 271, 13221–13227 (1996).
Yu, H., Kortylewski, M. & Pardoll, D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7, 41–51 (2007).
Hillmer, E. J., Zhang, H., Li, H. S. & Watowich, S. S. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 31, 1–15 (2016).
Xiao, L. et al. IL-9/STAT3/fatty acid oxidation-mediated lipid peroxidation contributes to Tc9 cell longevity and enhanced antitumor activity. J. Clin. Invest. 132, e153247 (2022).
MacDonagh, L. et al. BBI608 inhibits cancer stemness and reverses cisplatin resistance in NSCLC. Cancer Lett. 428, 117–126 (2018).
Bi, S. et al. Napabucasin (BBI608) eliminate AML cells in vitro and in vivo via inhibition of Stat3 pathway and induction of DNA damage. Eur. J. Pharm. 855, 252–261 (2019).
Bitsch, R. et al. STAT3 inhibitor Napabucasin abrogates MDSC immunosuppressive capacity and prolongs survival of melanoma-bearing mice. J. Immunother. Cancer 10, e004384 (2022).
Jonker, D. J. et al. Napabucasin versus placebo in refractory advanced colorectal cancer: a randomised phase 3 trial. Lancet Gastroenterol. Hepatol. 3, 263–270 (2018).
Kim, M. J. et al. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 335, 145–152 (2013).
Brambilla, L., Lahiri, T., Cammer, M. & Levy, D. E. STAT3 inhibitor OPB-51602 is cytotoxic to tumor cells through inhibition of complex I and ROS induction. iScience 23, 101822 (2020).
Hirpara, J. et al. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 25, 101076 (2019).
Genini, D. et al. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl Acad. Sci. USA 114, E4924–e4933 (2017).
Wong, A. L. et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 26, 998–1005 (2015).
Kasembeli, M. M. et al. TTI-101: a competitive inhibitor of STAT3 that spares oxidative phosphorylation and reverses mechanical allodynia in mouse models of neuropathic pain. Biochem. Pharm. 192, 114688 (2021).
Zhao, Y. et al. Analysis and validation of human targets and treatments using a hepatocellular carcinoma-immune humanized mouse model. Hepatology 74, 1395–1410 (2021).
Jung, K. H. et al. Multifunctional effects of a small-molecule STAT3 inhibitor on NASH and hepatocellular carcinoma in mice. Clin. Cancer Res. 23, 5537–5546 (2017).
Di, J. X. & Zhang, H. Y. C188-9, a small-molecule STAT3 inhibitor, exerts an antitumor effect on head and neck squamous cell carcinoma. Anticancer Drugs 30, 846–853 (2019).
Pedroza, M. et al. Role of STAT3 in skin fibrosis and transforming growth factor beta signalling. Rheumatology 57, 1838–1850 (2018).
Robinson, P. et al. Genetic and small-molecule modulation of stat3 in a mouse model of Crohn’s disease. J. Clin. Med. 11, 7020 (2022).
Sheng, W. et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 24, 1387–1402 (2014).
Luo, F. et al. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J. Immunother. Cancer 7, 245 (2019).
Guha, P. et al. STAT3 inhibition induces Bax-dependent apoptosis in liver tumor myeloid-derived suppressor cells. Oncogene 38, 533–548 (2019).
Pei, J. et al. STAT3 inhibition enhances CDN-induced STING signaling and antitumor immunity. Cancer Lett. 450, 110–122 (2019).
Moreira, D. et al. STAT3 inhibition combined with CpG immunostimulation activates antitumor immunity to eradicate genetically distinct castration-resistant prostate cancers. Clin. Cancer Res. 24, 5948–5962 (2018).
Jamieson, S., Wallace, C. E., Das, N., Bhattacharyya, P. & Bishayee, A. Guava (Psidium guajava L.): a glorious plant with cancer preventive and therapeutic potential. Crit. Rev. Food Sci. Nutr. 63, 192–223 (2023).
Sun, Q. et al. Flavonoids regulate tumor-associated macrophages - from structure-activity relationship to clinical potential (Review). Pharm. Res. 184, 106419 (2022).
Kalick, L. S. et al. Mangosteen for malignancy prevention and intervention: current evidence, molecular mechanisms, and future perspectives. Pharm. Res. 188, 106630 (2023).
Yang, J., Wang, L., Guan, X. & Qin, J. J. Inhibiting STAT3 signaling pathway by natural products for cancer prevention and therapy: In vitro and in vivo activity and mechanisms of action. Pharm. Res. 182, 106357 (2022).
Mahata, S. et al. In-silico and in-vitro investigation of STAT3-PIM1 heterodimeric complex: Its mechanism and inhibition by curcumin for cancer therapeutics. Int. J. Biol. Macromol. 208, 356–366 (2022).
Patel, S. S. et al. Cellular and molecular mechanisms of curcumin in prevention and treatment of disease. Crit. Rev. Food Sci. Nutr. 60, 887–939 (2020).
Tian, M., Tian, D., Qiao, X., Li, J. & Zhang, L. Modulation of Myb-induced NF-kB -STAT3 signaling and resulting cisplatin resistance in ovarian cancer by dietary factors. J. Cell Physiol. 234, 21126–21134 (2019).
Chen, Y. C. et al. Myricetin inhibits interferon-γ-induced PD-L1 and IDO1 expression in lung cancer cells. Biochem. Pharm. 197, 114940 (2022).
Liu, K. et al. Focus on immune checkpoint PD-1/PD-L1 pathway: new advances of polyphenol phytochemicals in tumor immunotherapy. Biomed. Pharmacother. 154, 113618 (2022).
Runtsch, M. C. et al. Itaconate and itaconate derivatives target JAK1 to suppress alternative activation of macrophages. Cell Metab. 34, 487–501.e488 (2022).
Guan, X. et al. Dual inhibition of MYC and SLC39A10 by a novel natural product STAT3 inhibitor derived from Chaetomium globosum suppresses tumor growth and metastasis in gastric cancer. Pharm. Res. 189, 106703 (2023).
Cianciulli, A., Calvello, R., Porro, C., Trotta, T. & Panaro, M. A. Understanding the role of SOCS signaling in neurodegenerative diseases: current and emerging concepts. Cytokine Growth Factor Rev. 37, 67–79 (2017).
Ahmed, C. M., Larkin, J. 3rd & Johnson, H. M. SOCS1 mimetics and antagonists: a complementary approach to positive and negative regulation of immune function. Front. Immunol. 6, 183 (2015).
McLornan, D. P., Pope, J. E., Gotlib, J. & Harrison, C. N. Current and future status of JAK inhibitors. Lancet 398, 803–816 (2021).
Liu, C., Kieltyka, J., Fleischmann, R., Gadina, M. & O’Shea, J. J. A decade of JAK inhibitors: what have we learned and what may be the future? Arthritis Rheumatol. 73, 2166–2178 (2021).
De Vries, L. C. S., Wildenberg, M. E., De Jonge, W. J. & D’Haens, G. R. The future of janus kinase inhibitors in inflammatory bowel disease. J. Crohns Colitis 11, 885–893 (2017).
Verstovsek, S. et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 363, 1117–1127 (2010).
Rosmarin, D. et al. Ruxolitinib cream for treatment of vitiligo: a randomised, controlled, phase 2 trial. Lancet 396, 110–120 (2020).
Zeiser, R. et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N. Engl. J. Med. 382, 1800–1810 (2020).
Hurwitz, H. I. et al. Randomized, double-blind, phase ii study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J. Clin. Oncol. 33, 4039–4047 (2015).
Mesa, R. A. et al. Effect of ruxolitinib therapy on myelofibrosis-related symptoms and other patient-reported outcomes in COMFORT-I: a randomized, double-blind, placebo-controlled trial. J. Clin. Oncol. 31, 1285–1292 (2013).
Kiladjian, J. J. et al. Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol. 7, e226–e237 (2020).
Harrison, C. N. et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 4, e317–e324 (2017).
Roskoski, R. Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update. Pharm. Res. 152, 104609 (2020).
Vannucchi, A. M. et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 372, 426–435 (2015).
Verstovsek, S. et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 366, 799–807 (2012).
Passamonti, F. et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol. 18, 88–99 (2017).
Harrison, C. N. et al. Addition of navitoclax to ongoing ruxolitinib therapy for patients with myelofibrosis with progression or suboptimal response: phase ii safety and efficacy. J. Clin. Oncol. 40, 1671–1680 (2022).
Dao, K. T. et al. Efficacy of ruxolitinib in patients with chronic neutrophilic leukemia and atypical chronic myeloid leukemia. J. Clin. Oncol. 38, 1006–1018 (2020).
Harrison, C. et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 366, 787–798 (2012).
Tefferi, A., Litzow, M. R. & Pardanani, A. Long-term outcome of treatment with ruxolitinib in myelofibrosis. N. Engl. J. Med. 365, 1455–1457 (2011).
Harrison, C. N. et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 30, 1701–1707 (2016).
Kvasnicka, H. M. et al. Long-term effects of ruxolitinib versus best available therapy on bone marrow fibrosis in patients with myelofibrosis. J. Hematol. Oncol. 11, 42 (2018).
Zeiser, R. et al. Ruxolitinib for glucocorticoid-refractory chronic graft-versus-host disease. N. Engl. J. Med. 385, 228–238 (2021).
Rudra, S. et al. Ruxolitinib: targeted approach for treatment of autoinflammatory very early onset inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 20, 1408–1410.e1402 (2022).
Bader-Meunier, B. et al. Effectiveness and safety of ruxolitinib for the treatment of refractory systemic idiopathic juvenile arthritis like associated with interstitial lung disease: a case report. Ann. Rheum. Dis. 81, e20 (2022).
Han, M. K. et al. Ruxolitinib in addition to standard of care for the treatment of patients admitted to hospital with COVID-19 (RUXCOVID): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Rheumatol. 4, e351–e361 (2022).
Fisher, D. A. C. et al. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFκB signaling. Leukemia 33, 1978–1995 (2019).
Wathes, R., Moule, S. & Milojkovic, D. Progressive multifocal leukoencephalopathy associated with ruxolitinib. N. Engl. J. Med. 369, 197–198 (2013).
Coltro, G. et al. A life-threatening ruxolitinib discontinuation syndrome. Am. J. Hematol. 92, 833–838 (2017).
Tvorogov, D. et al. Accumulation of JAK activation loop phosphorylation is linked to type I JAK inhibitor withdrawal syndrome in myelofibrosis. Sci. Adv. 4, eaat3834 (2018).
Houthuys, J. F., Wilmer, A. P., Peetermans, M., Meersseman, P. & Devos, T. Severe ARDS due to ruxolitinib discontinuation syndrome: case presentation and literature review. Heliyon 8, e11782 (2022).
Jagasia, M. et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label phase 2 trial. Blood 135, 1739–1749 (2020).
Guglielmelli, P. et al. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood 123, 2157–2160 (2014).
Passamonti, F. et al. Impact of ruxolitinib on the natural history of primary myelofibrosis: a comparison of the DIPSS and the COMFORT-2 cohorts. Blood 123, 1833–1835 (2014).
Verstovsek, S. et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J. Hematol. Oncol. 10, 156 (2017).
Verstovsek, S. et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J. Hematol. Oncol. 10, 55 (2017).
Marconi, V. C. et al. Randomized trial of ruxolitinib in antiretroviral-treated adults with human immunodeficiency virus. Clin. Infect. Dis. 74, 95–104 (2022).
Scott, L. J. Tofacitinib: a review of its use in adult patients with rheumatoid arthritis. Drugs 73, 857–874 (2013).
Dhillon, S. Tofacitinib: a review in rheumatoid arthritis. Drugs 77, 1987–2001 (2017).
Guimarães, P. O. et al. Tofacitinib in patients hospitalized with Covid-19 pneumonia. N. Engl. J. Med. 385, 406–415 (2021).
Sandborn, W. J. et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 376, 1723–1736 (2017).
Sandborn, W. J. et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N. Engl. J. Med. 367, 616–624 (2012).
Ytterberg, S. R. et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N. Engl. J. Med. 386, 316–326 (2022).
Wollenhaupt, J. et al. Safety and efficacy of tofacitinib, an oral janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, longterm extension studies. J. Rheumatol. 41, 837–852 (2014).
Kremer, J. M. et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 60, 1895–1905 (2009).
Sandborn, W. J. et al. Efficacy and safety of extended induction with tofacitinib for the treatment of ulcerative colitis. Clin. Gastroenterol. Hepatol. 20, 1821–1830.e1823 (2022).
Sandborn, W. J. et al. Efficacy and safety of tofacitinib in ulcerative colitis based on prior tumor necrosis factor inhibitor failure status. Clin. Gastroenterol. Hepatol. 20, 591–601.e598 (2022).
Sandborn, W. J. et al. Safety of tofacitinib for treatment of ulcerative colitis, based on 4.4 years of data from global clinical trials. Clin. Gastroenterol. Hepatol. 17, 1541–1550 (2019).
Gladman, D. et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N. Engl. J. Med. 377, 1525–1536 (2017).
Khosrow-Khavar, F., Kim, S. C., Lee, H., Lee, S. B. & Desai, R. J. Tofacitinib and risk of cardiovascular outcomes: results from the safety of TofAcitinib in routine care patients with Rheumatoid Arthritis (STAR-RA) study. Ann. Rheum. Dis. 81, 798–804 (2022).
Deodhar, A. et al. Tofacitinib for the treatment of ankylosing spondylitis: a phase III, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 80, 1004–1013 (2021).
Krueger, J. et al. Tofacitinib attenuates pathologic immune pathways in patients with psoriasis: a randomized phase 2 study. J. Allergy Clin. Immunol. 137, 1079–1090 (2016).
Sandborn, W. J. et al. A phase 2 study of tofacitinib, an oral Janus kinase inhibitor, in patients with Crohn’s disease. Clin. Gastroenterol. Hepatol. 12, 1485–1493.e1482 (2014).
Bissonnette, R. et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br. J. Dermatol. 175, 902–911 (2016).
Ma, C. et al. Innovations in oral therapies for inflammatory bowel disease. Drugs 79, 1321–1335 (2019).
Colombel, J. F. et al. Maintenance of remission with tofacitinib therapy in patients with ulcerative colitis. Clin. Gastroenterol. Hepatol. 20, 116–125.e115 (2022).
Kotwani, P., Terdiman, J. & Lewin, S. Tofacitinib for rescue therapy in acute severe ulcerative colitis: a real-world experience. J. Crohns Colitis 14, 1026–1028 (2020).
Takeuchi, T. Cytokines and cytokine receptors as targets of immune-mediated inflammatory diseases-RA as a role model. Inflamm. Regen. 42, 35 (2022).
Smolen, J. S. et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann. Rheum. Dis. 82, 3–18 (2022).
Fleischmann, R. et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N. Engl. J. Med. 367, 495–507 (2012).
Mease, P. et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N. Engl. J. Med. 377, 1537–1550 (2017).
Panés, J. et al. Tofacitinib for induction and maintenance therapy of Crohn’s disease: results of two phase IIb randomised placebo-controlled trials. Gut 66, 1049–1059 (2017).
Sandborn, W. J. et al. Tofacitinib for the treatment of ulcerative colitis: an integrated summary of up to 7.8 years of safety data from the global clinical program. J. Crohns Colitis 17, 338–351 (2022).
Damsky, W. et al. Inhibition of type 1 immunity with tofacitinib is associated with marked improvement in longstanding sarcoidosis. Nat. Commun. 13, 3140 (2022).
You, H. et al. Tofacitinib as a possible treatment for skin thickening in diffuse cutaneous systemic sclerosis. Rheumatology 60, 2472–2477 (2021).
Khosrow-Khavar, F., Desai, R. J., Lee, H., Lee, S. B. & Kim, S. C. Tofacitinib and risk of malignancy: results from the safety of tofacitinib in routine care patients with rheumatoid arthritis (STAR-RA) study. Arthritis Rheumatol. 74, 1648–1659 (2022).
Miyawaki, H. et al. Long-term effects of the janus kinase 1/2 inhibitor ruxolitinib on pulmonary hypertension and the cardiac function in a patient with myelofibrosis. Intern. Med. 59, 229–233 (2020).
Karpov, A. A. et al. Inhibition of JAK1,2 prevents fibrotic remodeling of pulmonary vascular bed and improves outcomes in the rat model of chronic thromboembolic pulmonary hypertension. Int. J. Mol. Sci. 23, 15646 (2022).
Hoisnard, L. et al. Adverse events associated with JAK inhibitors in 126,815 reports from the WHO pharmacovigilance database. Sci. Rep. 12, 7140 (2022).
Genovese, M. C. et al. Baricitinib in patients with refractory rheumatoid arthritis. N. Engl. J. Med. 374, 1243–1252 (2016).
Taylor, P. C. et al. Safety of baricitinib for the treatment of rheumatoid arthritis over a median of 4.6 and up to 9.3 years of treatment: final results from long-term extension study and integrated database. Ann. Rheum. Dis. 81, 335–343 (2022).
Tanaka, Y. et al. Clinical outcomes in patients switched from adalimumab to baricitinib due to non-response and/or study design: phase III data in patients with rheumatoid arthritis. Ann. Rheum. Dis. 78, 890–898 (2019).
Smolen, J. S. et al. Patient-reported outcomes from a randomised phase III study of baricitinib in patients with rheumatoid arthritis and an inadequate response to biological agents (RA-BEACON). Ann. Rheum. Dis. 76, 694–700 (2017).
Lei, Y. et al. A multicenter blinded preclinical randomized controlled trial on Jak1/2 inhibition in MRL/MpJ-Fas(lpr) mice with proliferative lupus nephritis predicts low effect size. Kidney Int. 99, 1331–1341 (2021).
Wallace, D. J. et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 392, 222–231 (2018).
King, B. et al. Efficacy and safety of the oral Janus kinase inhibitor baricitinib in the treatment of adults with alopecia areata: Phase 2 results from a randomized controlled study. J. Am. Acad. Dermatol. 85, 847–853 (2021).
Simpson, E. L. et al. Baricitinib in patients with moderate-to-severe atopic dermatitis: results from a randomized monotherapy phase 3 trial in the United States and Canada (BREEZE-AD5). J. Am. Acad. Dermatol. 85, 62–70 (2021).
Guttman-Yassky, E. et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: a phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J. Am. Acad. Dermatol. 80, 913–921.e919 (2019).
Bieber, T. et al. Efficacy and safety of baricitinib in combination with topical corticosteroids in patients with moderate-to-severe atopic dermatitis with inadequate response, intolerance or contraindication to ciclosporin: results from a randomized, placebo-controlled, phase III clinical trial (BREEZE-AD4). Br. J. Dermatol. 187, 338–352 (2022).
Taylor, P. C. et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N. Engl. J. Med. 376, 652–662 (2017).
Kalil, A. C. et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N. Engl. J. Med. 384, 795–807 (2021).
King, B. et al. Two phase 3 trials of baricitinib for alopecia areata. N. Engl. J. Med. 386, 1687–1699 (2022).
Wolfe, C. R. et al. Baricitinib versus dexamethasone for adults hospitalised with COVID-19 (ACTT-4): a randomised, double-blind, double placebo-controlled trial. Lancet Respir. Med. 10, 888–899 (2022).
Ely, E. W. et al. Efficacy and safety of baricitinib plus standard of care for the treatment of critically ill hospitalised adults with COVID-19 on invasive mechanical ventilation or extracorporeal membrane oxygenation: an exploratory, randomised, placebo-controlled trial. Lancet Respir. Med. 10, 327–336 (2022).
Marconi, V. C. et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 9, 1407–1418 (2021).
Stebbing, J. et al. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 20, 400–402 (2020).
Richardson, P. et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 395, e30–e31 (2020).
Cantini, F. et al. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J. Infect. 81, 318–356 (2020).
Cantini, F. et al. Beneficial impact of Baricitinib in COVID-19 moderate pneumonia; multicentre study. J. Infect. 81, 647–679 (2020).
Taylor, P. C. et al. Cardiovascular safety during treatment with baricitinib in rheumatoid arthritis. Arthritis Rheumatol. 71, 1042–1055 (2019).
Markham, A. & Keam, S. J. Peficitinib: first global approval. Drugs 79, 887–891 (2019).
Lee, Y. H. et al. Comparative efficacy and safety of peficitinib 25, 50, 100, and 150 mg in patients with active rheumatoid arthritis: a bayesian network meta-analysis of randomized controlled trials. Clin. Drug Investig. 40, 65–72 (2020).
Dhillon, S. Delgocitinib: first approval. Drugs 80, 609–615 (2020).
Worm, M. et al. The pan-JAK inhibitor delgocitinib in a cream formulation demonstrates dose response in chronic hand eczema in a 16-week randomized phase IIb trial. Br. J. Dermatol. 187, 42–51 (2022).
Worm, M. et al. Efficacy and safety of topical delgocitinib in patients with chronic hand eczema: data from a randomized, double-blind, vehicle-controlled phase IIa study. Br. J. Dermatol. 182, 1103–1110 (2020).
Nakagawa, H. et al. Long-term safety and efficacy of delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with atopic dermatitis. J. Dermatol. 47, 114–120 (2020).
Nakagawa, H. et al. Delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with moderate to severe atopic dermatitis: A phase 3, randomized, double-blind, vehicle-controlled study and an open-label, long-term extension study. J. Am. Acad. Dermatol. 82, 823–831 (2020).
Nakagawa, H. et al. Delgocitinib ointment in pediatric patients with atopic dermatitis: a phase 3, randomized, double-blind, vehicle-controlled study and a subsequent open-label, long-term study. J. Am. Acad. Dermatol. 85, 854–862 (2021).
Nakagawa, H. et al. Phase 2 clinical study of delgocitinib ointment in pediatric patients with atopic dermatitis. J. Allergy Clin. Immunol. 144, 1575–1583 (2019).
Garrido-Trigo, A. & Salas, A. Molecular structure and function of janus kinases: implications for the development of inhibitors. J. Crohns Colitis 14, S713–s724 (2020).
Nielsen, O. H., Boye, T. L., Chakravarti, D. & Gubatan, J. Selective tyrosine kinase 2 inhibitors in inflammatory bowel disease. Trends Pharm. Sci. 43, 424–436 (2022).
Pardanani, A. et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J. Clin. Oncol. 29, 789–796 (2011).
Pardanani, A. et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 1, 643–651 (2015).
Harrison, C. N. et al. Fedratinib in patients with myelofibrosis previously treated with ruxolitinib: an updated analysis of the JAKARTA2 study using stringent criteria for ruxolitinib failure. Am. J. Hematol. 95, 594–603 (2020).
Pardanani, A. et al. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 5, e335 (2015).
Harrison, C. N. et al. Safety and efficacy of fedratinib, a selective oral inhibitor of Janus kinase-2 (JAK2), in patients with myelofibrosis and low pretreatment platelet counts. Br. J. Haematol. 198, 317–327 (2022).
Gerds, A. T. et al. Myeloproliferative neoplasms, version 3.2022, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 20, 1033–1062 (2022).
Conigliaro, P. et al. Challenges in the treatment of rheumatoid arthritis. Autoimmun. Rev. 18, 706–713 (2019).
Mahajan, S. et al. VX-509 (decernotinib) is a potent and selective janus kinase 3 inhibitor that attenuates inflammation in animal models of autoimmune disease. J. Pharm. Exp. Ther. 353, 405–414 (2015).
Fleischmann, R. M. et al. A randomized, double-blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheumatol. 67, 334–343 (2015).
Genovese, M. C., van Vollenhoven, R. F., Pacheco-Tena, C., Zhang, Y. & Kinnman, N. VX-509 (decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol. 68, 46–55 (2016).
Genovese, M. C., Yang, F., Østergaard, M. & Kinnman, N. Efficacy of VX-509 (decernotinib) in combination with a disease-modifying antirheumatic drug in patients with rheumatoid arthritis: clinical and MRI findings. Ann. Rheum. Dis. 75, 1979–1983 (2016).
Etra, A. et al. Effective treatment of low-risk acute GVHD with itacitinib monotherapy. Blood 141, 481–489 (2023).
Zeiser, R. et al. Efficacy and safety of itacitinib versus placebo in combination with corticosteroids for initial treatment of acute graft-versus-host disease (GRAVITAS-301): a randomised, multicentre, double-blind, phase 3 trial. Lancet Haematol. 9, e14–e25 (2022).
Burmester, G. R. et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 391, 2503–2512 (2018).
Duggan, S. & Keam, S. J. Upadacitinib: first approval. Drugs 79, 1819–1828 (2019).
Kavanaugh, A. et al. Safety and efficacy of filgotinib: up to 4-year results from an open-label extension study of phase II rheumatoid arthritis programs. J. Rheumatol. 48, 1230–1238 (2021).
Smolen, J. S. et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet 393, 2303–2311 (2019).
Deodhar, A. et al. Upadacitinib for the treatment of active non-radiographic axial spondyloarthritis (SELECT-AXIS 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 400, 369–379 (2022).
Danese, S. et al. Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double-blind, randomised trials. Lancet 399, 2113–2128 (2022).
Gooderham, M. J. et al. Efficacy and safety of oral janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: a phase 2 randomized clinical trial. JAMA Dermatol. 155, 1371–1379 (2019).
Ständer, S. et al. High threshold efficacy responses in moderate-to-severe atopic dermatitis are associated with additional quality of life benefits: pooled analyses of abrocitinib monotherapy studies in adults and adolescents. J. Eur. Acad. Dermatol. Venereol. 36, 1308–1317 (2022).
Silverberg, J. I. et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 156, 863–873 (2020).
Eichenfield, L. F. et al. Efficacy and safety of abrocitinib in combination with topical therapy in adolescents with moderate-to-severe atopic dermatitis: the JADE TEEN randomized clinical trial. JAMA Dermatol. 157, 1165–1173 (2021).
Shi, V. Y. et al. Phase 3 efficacy and safety of abrocitinib in adults with moderate-to-severe atopic dermatitis after switching from dupilumab (JADE EXTEND). J. Am. Acad. Dermatol. 87, 351–358 (2022).
Napolitano, M. et al. The efficacy and safety of abrocitinib as a treatment option for atopic dermatitis: a short report of the clinical data. Drug Des. Devel. Ther. 15, 1135–1147 (2021).
Reich, K. et al. Magnitude and time course of response to abrocitinib for moderate-to-severe atopic dermatitis. J. Allergy Clin. Immunol. Pract. 10, e3222 (2022).
Simpson, E. L. et al. Integrated safety analysis of abrocitinib for the treatment of moderate-to-severe atopic dermatitis from the phase II and phase III clinical trial program. Am. J. Clin. Dermatol. 22, 693–707 (2021).
Chimalakonda, A. et al. Selectivity profile of the tyrosine kinase 2 inhibitor deucravacitinib compared with janus kinase 1/2/3 inhibitors. Dermatol. Ther. 11, 1763–1776 (2021).
Hoy, S. M. Deucravacitinib: first approval. Drugs 82, 1671–1679 (2022).
Armstrong, A. W. et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J. Am. Acad. Dermatol. 88, 29–39 (2023).
Strober, B. et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 Program fOr Evaluation of TYK2 inhibitor psoriasis second trial. J. Am. Acad. Dermatol. 88, 40–51 (2023).
Morand, E. et al. Deucravacitinib, a tyrosine kinase 2 inhibitor, in systemic lupus erythematosus: a phase ii, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 75, 242–252 (2023).
Chen, B. et al. Efficacy and safety of ivarmacitinib in patients with moderate-to-severe, active, ulcerative colitis: a phase II study. Gastroenterology 163, 1555–1568 (2022).
Wu, H. et al. JAK1-STAT3 blockade by JAK inhibitor SHR0302 attenuates inflammatory responses of adjuvant-induced arthritis rats and decreases Th17 and total B cells. Jt. Bone Spine 83, 525–532 (2016).
Sun, X. et al. Preventive and therapeutic effects of a novel JAK inhibitor SHR0302 in acute graft-versus-host disease. Cell Transpl. 30, 9636897211033778 (2021).
Zhao, Y. et al. Efficacy and safety of SHR0302, a highly selective janus kinase 1 inhibitor, in patients with moderate to severe atopic dermatitis: a phase II randomized clinical trial. Am. J. Clin. Dermatol. 22, 877–889 (2021).
Su, Q. et al. Discovery of (2R)-N-[3-[2-[(3-Methoxy-1-methyl-pyrazol-4-yl)amino]pyrimidin-4-yl]-1H-indol-7-yl]-2-(4-methylpiperazin-1-yl)propenamide (AZD4205) as a potent and selective janus kinase 1 inhibitor. J. Med. Chem. 63, 4517–4527 (2020).
Tefferi, A., Gangat, N. & Pardanani, A. Jaktinib (JAK1/2 inhibitor): a momelotinib derivative with similar activity and optimized dosing schedule. Am. J. Hematol. 97, 1507–1509 (2022).
Liu, J. et al. A phase i, randomized, double-blind, placebo-controlled, single ascending dose, multiple ascending dose and food effect study to evaluate the tolerance, pharmacokinetics of jaktinib, a new selective janus kinase inhibitor in healthy chinese volunteers. Front. Pharm. 11, 604314 (2020).
Zhang, Y. et al. Safety and efficacy of jaktinib in the treatment of Janus kinase inhibitor-naïve patients with myelofibrosis: results of a phase II trial. Am. J. Hematol. 97, 1510–1519 (2022).
Meng, X. et al. Potential for jaktinib hydrochloride to treat cytokine storms in patients with COVID-19. Biosci. Trends 14, 161–167 (2020).
Philips, R. L. et al. The JAK-STAT pathway at 30: much learned, much more to do. Cell 185, 3857–3876 (2022).
Thoidingjam, L. K. et al. Small Molecule Inhibitors of Interferon-Induced JAK-STAT Signalling. Angew. Chem. Int. Ed. Engl. 61, e202205231 (2022).
Pohóczky, K. et al. Discovery of novel targets in a complex regional pain syndrome mouse model by transcriptomics: TNF and JAK-STAT pathways. Pharm. Res. 182, 106347 (2022).
Patterson, H., Nibbs, R., McInnes, I. & Siebert, S. Protein kinase inhibitors in the treatment of inflammatory and autoimmune diseases. Clin. Exp. Immunol. 176, 1–10 (2014).
Choy, E. H. Clinical significance of Janus Kinase inhibitor selectivity. Rheumatology 58, 953–962 (2019).
Lin, C. M., Cooles, F. A. & Isaacs, J. D. Basic mechanisms of JAK inhibition. Mediterr. J. Rheumatol. 31, 100–104 (2020).
Cook, N., Hansen, A. R., Siu, L. L. & Abdul Razak, A. R. Early phase clinical trials to identify optimal dosing and safety. Mol. Oncol. 9, 997–1007 (2015).
Pérez-Jeldres, T. et al. Targeting cytokine signaling and lymphocyte traffic via small molecules in inflammatory bowel disease: JAK inhibitors and S1PR agonists. Front. Pharm. 10, 212 (2019).
Sabino, J., Verstockt, B., Vermeire, S. & Ferrante, M. New biologics and small molecules in inflammatory bowel disease: an update. Ther. Adv. Gastroenterol. 12, 1756284819853208 (2019).
Chen, C. X. et al. JAK-inhibitors for coronavirus disease-2019 (COVID-19): a meta-analysis. Leukemia 35, 2616–2620 (2021).
Kleppe, M. et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell 33, 29–43.e27 (2018).
Pastore, F. et al. PRMT5 inhibition modulates E2F1 methylation and gene-regulatory networks leading to therapeutic efficacy in JAK2(V617F)-mutant MPN. Cancer Discov. 10, 1742–1757 (2020).
Blair, H. A. Fedratinib: first approval. Drugs 79, 1719–1725 (2019).
Talpaz, M. & Kiladjian, J. J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 35, 1–17 (2021).
Zhang, W. et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J. Natl. Cancer Inst. 100, 184–198 (2008).
Fridman, J. S. et al. Preclinical evaluation of local JAK1 and JAK2 inhibition in cutaneous inflammation. J. Invest. Dermatol. 131, 1838–1844 (2011).
Al-Bawardy, B., Shivashankar, R. & Proctor, D. D. Novel and emerging therapies for inflammatory bowel disease. Front. Pharm. 12, 651415 (2021).
Acknowledgements
This work was funded by the National Key Research and Development Program of China (2021YFC2301800), and the National Nature Science Foundation of China (82200673 and U20A20343).
Author information
Authors and Affiliations
Contributions
L.L. and J.L. designed the study, and reviewed and edited the manuscript; C.X., Q.Y., and X.G. participated in the original draft preparation; Q.S., X.Y., Q.C., and Z.B. collected the references and help with reviewing the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xue, C., Yao, Q., Gu, X. et al. Evolving cognition of the JAK-STAT signaling pathway: autoimmune disorders and cancer. Sig Transduct Target Ther 8, 204 (2023). https://doi.org/10.1038/s41392-023-01468-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01468-7
Subjects
This article is cited by
-
Intricate effects of post-translational modifications in liver cancer: mechanisms to clinical applications
Journal of Translational Medicine (2024)
-
Signaling pathways activated and regulated by stem cell-derived exosome therapy
Cell & Bioscience (2024)
-
Activation of kappa opioid receptor suppresses post-traumatic osteoarthritis via sequestering STAT3 on the plasma membrane
Cell Communication and Signaling (2024)
-
JAK/STAT in leukemia: a clinical update
Molecular Cancer (2024)
-
Writers, readers, and erasers RNA modifications and drug resistance in cancer
Molecular Cancer (2024)