
Abstract 抽象
Single-cell analyses parse the brain’s billions of neurons into thousands of ‘cell-type’ clusters residing in different brain structures1. Many cell types mediate their functions through targeted long-distance projections allowing interactions between specific cell types. Here we used epi-retro-seq2 to link single-cell epigenomes and cell types to long-distance projections for 33,034 neurons dissected from 32 different regions projecting to 24 different targets (225 source-to-target combinations) across the whole mouse brain. We highlight uses of these data for interrogating principles relating projection types to transcriptomics and epigenomics, and for addressing hypotheses about cell types and connections related to genetics. We provide an overall synthesis with 926 statistical comparisons of discriminability of neurons projecting to each target for every source. We integrate this dataset into the larger BRAIN Initiative Cell Census Network atlas, composed of millions of neurons, to link projection cell types to consensus clusters. Integration with spatial transcriptomics further assigns projection-enriched clusters to smaller source regions than the original dissections. We exemplify this by presenting in-depth analyses of projection neurons from the hypothalamus, thalamus, hindbrain, amygdala and midbrain to provide insights into properties of those cell types, including differentially expressed genes, their associated cis-regulatory elements and transcription-factor-binding motifs, and neurotransmitter use.
单细胞分析将大脑的数十亿个神经元解析为位于不同大脑结构中的数千个“细胞型”簇1。许多细胞类型通过靶向长距离投射介导其功能,从而允许特定细胞类型之间的相互作用。在这里,我们使用 epi-retro-seq2 将单细胞表观基因组和细胞类型与从 32 个不同区域解剖的 33,034 个神经元的长距离投影联系起来,投射到整个小鼠大脑的 24 个不同靶点(225 个源到靶点组合)。我们重点介绍了这些数据的用途,用于询问将投影类型与转录组学和表观基因组学相关的原则,以及解决与遗传学相关的细胞类型和连接的假设。我们提供了一个整体综合,其中包含 926 个统计比较,比较了每个来源投射到每个目标的神经元的判别能力。我们将此数据集集成到更大的 BRAIN Initiative 细胞普查网络图谱中,该图谱由数百万个神经元组成,以将投影细胞类型与共有聚类联系起来。与空间转录组学的集成进一步将投影丰富的簇分配给比原始解剖更小的源区域。我们通过对来自下丘脑、丘脑、后脑、杏仁核和中脑的投射神经元进行深入分析来举例说明这一点,以提供对这些细胞类型特性的见解,包括差异表达基因、它们相关的顺式调节元件和转录因子结合基序,以及神经递质的使用。
Similar content being viewed by others
其他人正在查看类似内容


Main 主要
In any given brain, each neuron contributes uniquely to brain function. Nevertheless, neurons can be grouped into types on the basis of similarities and differences across several dimensions, including epigenetic state, gene expression, anatomy and physiology. Single-cell genomic technologies have been particularly impactful for cell-type classification owing to their high throughput (millions of cells assayed) and dimensionality (thousands of genes and even more genetic loci) leading to the identification of large numbers of transcriptomic and epigenomic clusters corresponding to possible cell types across the entire mouse brain.
在任何给定的大脑中,每个神经元都对大脑功能做出独特的贡献。然而,神经元可以根据多个维度的相似性和差异性进行分组,包括表观遗传状态、基因表达、解剖学和生理学。单细胞基因组技术因其高通量(检测数百万个细胞)和维度(数千个基因甚至更多的遗传位点)而对细胞类型分类特别有影响,从而可以识别出与整个小鼠大脑中可能的细胞类型相对应的大量转录组和表观基因组簇。
A prominent and distinguishing anatomical feature of many brain neuron types is their long-distance axonal projections. Long-distance projections can be directly related to single-neuron gene expression or epigenomes by use of powerful linking technologies, including Barcoded Anatomy Resolved by Sequencing (BARseq)3,4, retro-seq5,6 and epi-retro-seq2. Previous studies have used retro-seq and epi-retro-seq to link mouse neocortical2,5,6, hypothalamic7 and thalamic projection cell types8 to their genetic and epigenetic clusters, revealing complex but predictable relationships. For example, cortical neurons projecting solely to intratelencephalic (IT) targets fall into different clusters compared with those that project to extratelencephalic (ET) targets. By contrast, cortical layer 2/3 IT neuron types projecting to different cortical areas typically co-cluster despite having quantifiable and predictable genetic and epigenetic differences across the populations2,6. In the face of this complexity, it is unclear how single-cell genetic and epigenetic assays can be used to inform the structure and function of brain cell types and how neuronal structure can predict genetics and epigenetics. Further, it is unclear whether the principles learned from more limited previous studies can be extended to the entire brain, or whether there are different principles linking projection status to epigenetics for different brain areas.
许多脑神经元类型的一个突出和显着的解剖特征是它们的长距离轴突投射。通过使用强大的连接技术,包括通过测序分辨的条形码解剖学 (BARseq)3,4、retro-seq5,6 和 epi-retro-seq2,长距离投影可以与单神经元基因表达或表观基因组直接相关。以前的研究使用 retro-seq 和 epi-retro-seq 将小鼠新皮层 2,5,6、下丘脑7 和丘脑投射细胞类型8 与它们的遗传和表观遗传簇联系起来,揭示了复杂但可预测的关系。例如,与投射到端外 (ET) 目标的皮层神经元相比,仅投射到端脑内 (IT) 目标的皮层神经元属于不同的集群。相比之下,投射到不同皮层区域的皮层 2/3 IT 神经元类型通常共聚集,尽管群体之间存在可量化和可预测的遗传和表观遗传差异2,6。面对这种复杂性,目前尚不清楚如何使用单细胞遗传和表观遗传学分析来告知脑细胞类型的结构和功能,以及神经元结构如何预测遗传学和表观遗传学。此外,目前尚不清楚从更有限的先前研究中学到的原理是否可以扩展到整个大脑,或者是否存在不同的原理将投射状态与不同大脑区域的表观遗传学联系起来。
To address these questions, we used epi-retro-seq to assay 33,034 neurons from 225 source-to-target combinations across the entire mouse brain. This approach combines retrograde labelling with single-nucleus methylation sequencing (snmC-seq), which allows identification of potential gene regulatory elements and prediction of gene expression in the same neuron. Gene expression can be predicted because non-CG (CH; in which H represents A, T or C) methylation of gene bodies is inversely related to RNA expression9,10, and epigenetic elements regulating expression can be identified using methylation at CG (mCG) dinucleotides9. It is also expected that epi-retro-seq can provide unique insight into developmental mechanisms that shape connectivity because CH methylation accumulates during and peaks at the end of the developmental critical period, and mCG is reconfigured during synaptic development11.
为了回答这些问题,我们使用 epi-retro-seq 分析了整个小鼠大脑中来自 225 个源到靶组合的 33,034 个神经元。这种方法将逆行标记与单核甲基化测序 (snmC-seq) 相结合,从而可以识别潜在的基因调控元件并预测同一神经元中的基因表达。基因表达可以预测,因为基因体的非 CG(CH;其中 H 代表 A、T 或 C)甲基化与 RNA 表达呈负相关 9,10,并且可以通过 CG (mCG) 二核苷酸的甲基化来鉴定调节表达的表观遗传元件9。还预计 epi-retro-seq 可以为塑造连接的发育机制提供独特的见解,因为 CH 甲基化在发育关键期积累并在发育关键期结束时达到峰值,并且 mCG 在突触发育期间重新配置11。
Epi-retro-seq of 225 projections
225 个投影的 Epi-retro-seq
To link single-neuron epigenomes to their projection targets and cell body locations, we used epi-retro-seq2. A retrogradely infecting AAV vector expressing Cre-recombinase (AAV-retro-Cre12) was injected into the brains of Cre-dependent, nuclear-GFP-expressing reporter mice (INTACT-Cre9) at a target region of interest (Fig. 1a). Four mice (two male and two female) were injected for each of twenty-four different target brain areas, including targets in the isocortex (CTX), hippocampal formation, olfactory areas, amygdala (AMY), cerebral nuclei (CNU), interbrain (IB), midbrain (MB), hindbrain (HB) and cerebellum (Fig. 1a,b and Supplementary Table 1). After 2 weeks, mice were killed and the brain was hand dissected into 32 possible source regions13 spanning the same major brain structures as the target injections (Fig. 1a,b and Extended Data Fig. 1). For any given mouse, dissected sources corresponding to locations with known projections to the target were selected for profiling. Nucleus preparations were made from dissected source tissue and subjected to fluorescence-activated nuclear sorting for GFP+NeuN+ retrogradely labelled neuronal nuclei that were then processed for snmC-seq14,15,16 (Fig. 1a and Methods).
为了将单神经元表观基因组与其投影靶点和细胞体位置联系起来,我们使用了 epi-retro-seq2。将表达 Cre 重组酶 (AAV-retro-Cre12) 的逆行感染 AAV 载体注射到 Cre 依赖性、表达核 GFP 的报告小鼠 (INTACT-Cre9) 的大脑中,位于感兴趣的目标区域(图 D)。1a). 为 24 个不同的目标大脑区域分别注射了四只小鼠(两只雄性和两只雌性),包括等皮层 (CTX)、海马形成、嗅觉区域、杏仁核 (AMY)、脑核 (CNU)、脑间 (IB)、中脑 (MB)、后脑 (HB) 和小脑的目标 (图 .1a、b 和补充表 1)。2 周后,杀死小鼠,将大脑手工解剖成 32 个可能的源区域13,跨越与目标注射相同的主要大脑结构(图 D)。1a,b 和扩展数据图对于任何给定的鼠标,选择与已知投射到目标的位置相对应的解剖源进行分析。细胞核制剂由解剖的源组织制成,并对 GFP+NeuN+ 逆行标记的神经元核进行荧光激活核分选,然后将其加工为 snmC-seq14、15、16(图 D)。1a 和方法)。
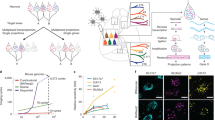
图 1:全脑投射神经元的表观基因组景观。

a, Schematics of the epi-retro-seq workflow for retrogradely labelling and epigenetically profiling single projection neurons. The three-dimensional brain contours of the source regions (top right) were derived from the Allen Mouse Brain Reference Atlas (http://atlas.brain-map.org, © 2017 Allen Institute for Brain Science, version 3 (2020)). The diagrams of the brain slices (top left) and the images of the surgical set up, microscope and library preparation apparatus in the workflow were created with BioRender.com. b, A total of 225 source–target combinations profiled using epi-retro-seq (blue dots) from 32 different source regions (rows) projecting to 24 different targets (columns) across the whole mouse brain. The colour palettes of sources (left), targets (bottom) and region groups (top and right) are labeled on the side and used across the article. c, Joint two-dimensional t-SNE of epi-retro-seq (n = 35,938) and unbiased snmC-seq (n = 276,187) neurons. snmC-seq neurons are shown in grey and epi-retro-seq neurons are coloured by cell subclass (top), the source regions of neurons (middle) or their projection targets (bottom; n = 33,034, after removing the experiments with less confident target assignment). d, As an example, AUROC for pairwise comparisons of AMY neurons projecting to nine targets. Higher AUROC scores suggest greater distinguishability between the compared projections based on their gene-body mCH levels. e, Joint t-SNE of whole-mouse-brain neurons from epi-retro-seq (n = 35,743), unbiased snmC-seq (n = 266,740) and scRNA-seq (n = 2,434,472) coloured by cell subclass. f, As an illustration, the proportion (prop.) of neurons found in each AMY cell cluster (row) that projects to each target (column). Only clusters that were enriched for projection neurons are shown and values are z-score normalized across targets. g, A sagittal brain slice for MERFISH with all neurons coloured by their assigned subclasses. h, An illustration of joint analysis of single-cell transcriptomes and DNA methylomes that enables the characterization of gene expression patterns of DEGs between these projection-enriched clusters, as well as the mCG levels of DEG-associated putative CREs, as marked by DMRs. i, An illustration of GRN linking TFs, CRE and target genes. AI, agranular insular cortex; AUD, auditory cortex; AUDp, primary auditory cortex; CAa, anterior cornu ammonis; CAp, posterior cornu ammonis; CB, cerebellum; CBN, cerebellar nuclei; CGE, caudal ganglionic eminence; CT, corticothalamic; DGa, anterior dentate gyrus; DGp, posterior dentate gyrus; GC, granule cell; GP, globus pallidus; HPF, hippocampal formation; IC, inferior colliculus; IO, inferior olivary; LSX, lateral septal complex; MGE, medial ganglionic eminence; MSN, medium spiny neurons; NP, near projecting; OLF, olfactory areas; PAG, periaqueductal grey; PIRa, anterior piriform cortex; PIRp, posterior piriform cortex; THl, anterior lateral thalamus; THm, anterior medial thalamus; THp, posterior TH; VIS, visual cortex.
a,用于逆行标记和表观遗传分析单个投射神经元的 epi-retro-seq 工作流程示意图。源区域的三维大脑轮廓(右上)来自艾伦小鼠大脑参考图谱(http://atlas.brain-map.org,2017 © 年艾伦脑科学研究所,第 3 版(2020 年))。工作流程中的脑切片图(左上)和手术装置、显微镜和文库制备装置的图像是使用 BioRender.com 创建的。b,使用 epi-retro-seq(蓝点)分析了总共 225 个源-靶点组合,这些组合来自 32 个不同的源区域(行),投射到整个小鼠大脑的 24 个不同目标(列)。来源(左)、目标(下)和区域组(上和右)的调色板在侧面标记,并在整篇文章中使用。c,epi-retro-seq (n = 35,938) 和无偏倚 snmC-seq (n = 276,187) 神经元的联合二维 t-SNE。snmC-seq 神经元以灰色显示,epi-retro-seq 神经元按细胞亚类(上)、神经元的源区域(中)或其投射目标(下)着色;n = 33,034,在去除目标分配置信度较低的实验后)。d,例如,AUROC 用于对投射到 9 个目标的 AMY 神经元进行成对比较。较高的 AUROC 分数表明基于基因体 mCH 水平的比较预测之间的可区分性更强。e,来自 epi-retro-seq (n = 35,743)、无偏倚 snmC-seq (n = 266,740) 和 scRNA-seq (n = 2,434,472) 的全小鼠脑神经元的联合 t-SNE,按细胞亚类着色。 f,例如,在投射到每个目标(列)的每个 AMY 细胞簇(行)中发现的神经元的比例(prop.)。仅显示为投影神经元而富集的集群,并且值在目标之间标准化 z 分数。g,MERFISH 的矢状脑切片,所有神经元都由其分配的亚类着色。h,单细胞转录组和 DNA 甲基化组联合分析的图示,能够表征这些投射丰富的簇之间 DEGs 的基因表达模式,以及 DMR 标记的 DEG 相关推定 CRE 的 mCG 水平。i,GRN 连接 TFs、CRE 和靶基因的图示。AI,无颗粒岛叶皮层;AUD,听觉皮层;AUDp,初级听觉皮层;CAa,前角氨;CAp,后山祸氨;CB, 小脑;CBN,小脑核;CGE,尾神经节隆起;CT,皮质丘脑;DGa,前齿状回;DGp,后齿状回;GC, 颗粒细胞;GP, 苍白球;HPF,海马形成;IC,下丘;IO,下橄榄;LSX,外侧间隔复合体;MGE,内侧神经节隆起;MSN,中等棘状神经元;NP,近突起;OLF,嗅觉区;PAG,导水管周围灰色;PIRa,前梨状皮层;PIRp,后梨状皮层;THl,前外侧丘脑;THm,前内侧丘脑;THp,后 TH;VIS,视觉皮层。
After basic quality control, we recovered 48,032 single-cell methylomes (Supplementary Table 2) that were mapped to an unbiased sample of snmC-seq data with 301,626 cells17 to carry out cell-type classification, and for removal of potential doublets (Methods and Extended Data Fig. 2a–e). Each single neuron in the epi-retro-seq sample was assigned to 1 of the 2,304 level 4 clusters identified in our companion study17. We have previously described cortical neurons from the same eight cortical sources included here and projecting to four cortical and six subcortical targets (63 combinations)2. For cortical sources, we now incorporate data for an additional five cortical targets and two more subcortical targets, with quality control steps similar to those in our previous work to eliminate experiments with inadvertent spread of injected AAV-retro-Cre into source regions (Methods). In total, 33,034 single-nucleus methylomes were analysed from 225 source-to-target combinations for which the projection target could be confidently assigned (Supplementary Table 3). These neurons were mapped to the unbiased snmC-seq dataset to visualize the epigenetic similarity of projection neurons across cell subclasses, sources and targets (Fig. 1c).
经过基本的质量控制,我们回收了 48,032 个单细胞甲基化组(补充表 2),这些细胞被映射到具有 301,626 个细胞17 的 snmC-seq 数据的无偏样本,以进行细胞类型分类,并去除潜在的双峰(方法和扩展数据图 2)。2a-e)。epi-retro-seq 样本中的每个单个神经元都被分配到我们的配套研究17 中确定的 2,304 个 4 级集群中的 1 个。我们之前已经描述了来自此处包含的相同 8 个皮层来源的皮层神经元,并投射到 4 个皮层和 6 个皮层下靶点(63 种组合)2。对于皮质来源,我们现在整合了另外五个皮质靶点和另外两个皮层下靶点的数据,其质量控制步骤类似于我们之前的工作,以消除注射的 AAV-retro-Cre 无意中扩散到源区域的实验(方法)。总共分析了来自 225 个源到目标组合的 33,034 个单核甲基化组,可以自信地分配其投射目标(补充表 3)。这些神经元被映射到无偏倚的 snmC-seq 数据集,以可视化投影神经元跨细胞亚类、来源和靶标的表观遗传相似性(图 .1c)。
Data analysis approaches across the brain
跨大脑的数据分析方法
Overarching questions that can be addressed by this large dataset include the distinguishability of neurons from a given source that project to different targets, and whether neurons in different sources that project to the same target combinations are more or less distinguishable. To provide a resource that can be used to address the distinguishability of neurons with different projection targets, we trained linear models to distinguish neurons projecting to pairs of different targets on the basis of DNA methylation, and quantify which projection types are more different than the others by computing the model performance through area under the curve of the receiver operating characteristic (AUROC) for each of the target pairs from every source region (926 pairwise comparisons in total; Fig. 1d, Methods, Extended Data Fig. 3 and Supplementary Table 4).
这个大型数据集可以解决的首要问题包括来自给定来源投射到不同目标的神经元的可区分性,以及投射到相同目标组合的不同来源中的神经元是否或多或少可区分。为了提供可用于解决具有不同投射目标的神经元的可区分性的资源,我们训练了线性模型,以根据 DNA 甲基化区分投射到不同目标对的神经元,并通过通过来自每个源区域的每个目标对的接收者操作特征曲线下面积 (AUROC) 计算模型性能来量化哪些投射类型比其他投影类型更不同 (926总计成对比较;无花果。1d, 方法, 扩展数据 图3 和补充表 4)。
To facilitate further, comprehensive multimodal characterization of projection neuron types, we integrated the epi-retro-seq data with unbiased samples of snmC-seq described above17, and single-cell RNA-seq (scRNA-seq) data for 2.6 million neurons from 87 micro-dissected brain regions18 (Fig. 1e, Methods and Extended Data Fig. 4). Alignment of epi-retro-seq data to these larger and carefully annotated datasets allows for the confident assignment of our cells to consensus clusters and enables the use of consistent nomenclature to describe the correspondence between projection targets and cell types or clusters. We carried out co-clustering of the three datasets to identify the cell clusters enriched in each projection type (Fig. 1f, Methods and Extended Data Fig. 5). It should be noted that in addition to clusters that are identified as being enriched in projection neurons, there are also neurons in other clusters without statistically significant enrichment. The absence of statistically significant enrichment should not be interpreted as an absence of projections from neurons belonging to a particular cluster.
为了促进投射神经元类型的进一步、全面的多模态表征,我们将 epi-retro-seq 数据与上述 snmC-seq 的无偏样本17 以及来自 87 个显微解剖大脑区域的 260 万个神经元的单细胞 RNA-seq (scRNA-seq) 数据整合在一起18(图 1)。1e,方法和扩展数据图4). 将 epi-retro-seq 数据与这些更大且仔细注释的数据集对齐,可以自信地将我们的细胞分配到共有集群,并能够使用一致的命名法来描述投影目标与细胞类型或集群之间的对应关系。我们对三个数据集进行了共聚类,以确定在每种投影类型中富集的细胞簇(图 D)。1f,方法和扩展数据图5). 应该注意的是,除了被鉴定为在投射神经元中富集的簇外,其他簇中还有神经元没有统计学意义的富集。没有统计学上显着的富集不应被解释为没有来自属于特定集群的神经元的投影。
Although microdissections effectively separate fairly small structures, most dissected source regions contain still smaller known anatomical regions, as typically illustrated in mouse brain atlases13. To potentially link projection-enriched clusters from particular sources to more precise anatomical loci, we carried out further integration with multiplexed error robust fluorescence in situ hybridization (MERFISH) data, allowing examination of the spatial locations of the cells belonging to particular clusters (Fig. 1g and Extended Data Fig. 6). Joint atlasing of single-neuron transcriptomes and epigenomes further allowed analyses of both the signature genes in projection-enriched clusters based on RNA expression, and methylation profiles to identify differentially methylated regions (DMRs) as putative cis-regulatory elements (CREs) and transcription factors (TFs) whose binding motifs are enriched in these DMRs (Fig. 1h). On the basis of the motif enrichment and the correlation between gene expression and DNA methylation, we constructed gene regulatory networks (GRNs) with TFs, DMRs and target genes as nodes (Fig. 1i). The GRNs allowed us to identify the most consistent changes across different data modalities, which pinpoint candidate regulators of projection-enriched clusters.
尽管显微解剖有效地分离了相当小的结构,但大多数解剖的源区域包含更小的已知解剖区域,如小鼠脑图谱13 中通常所示。为了有可能将来自特定来源的富含投影的簇与更精确的解剖位点联系起来,我们进一步整合了多路复用误差稳健荧光原位杂交 (MERFISH) 数据,从而可以检查属于特定簇的细胞的空间位置(图 D)。1g 和扩展数据图6). 单神经元转录组和表观基因组的联合图谱分析进一步允许基于 RNA 表达分析投射富集簇中的特征基因,以及甲基化谱,以识别差异甲基化区域 (DMR) 作为假定的顺式调节元件 (CRE) 和转录因子 (TFs),其结合基序在这些 DMR 中富集(图 D)。基于基序富集以及基因表达与 DNA 甲基化之间的相关性,我们构建了以 TFs、DMRs 和靶基因为节点的基因调控网络 (GRN) (图 .GRN 使我们能够识别不同数据模态中最一致的变化,从而确定投影丰富的集群的候选调节因子。
Extended Data Figs. 3–6 allow visualization of the integrative analysis approaches described above (for example, Fig. 1d–i) for all source-to-target combinations in our dataset. These integrative analyses were facilitated by combining source regions from the whole-brain datasets into 12 larger ‘region groups’ that were common to all 3 data modalities, before integration (Extended Data Fig. 2f,g). The groups include CTX, retro hippocampal region (RHP), piriform area (PIR), hippocampal region (HIP), main olfactory bulb and anterior olfactory nucleus (MOB + AON), striatum (STR), pallidum (PAL), AMY, thalamus (TH), hypothalamus (HY), MB and HB.
扩展数据图3-6 允许上述综合分析方法的可视化(例如,图 D)。1d-i)对于我们数据集中的所有源到目标组合。在整合之前,通过将全脑数据集中的源区域组合成 12 个更大的“区域组”来促进这些整合分析,这些区域组是所有 3 种数据模式共有的(扩展数据图 .2f,g)。这些组包括 CTX、后海马区 (RHP)、梨状区 (PIR)、海马区 (HIP)、主嗅球和前嗅核 (MOB + AON)、纹状体 (STR)、梅毒球 (PAL)、AMY、丘脑 (TH)、下丘脑 (HY)、MB 和 HB。
Below, we focus on a subset of all possible analyses of this very large dataset to highlight the utility of the data and to provide examples of interest. To facilitate further analyses of the complete dataset, we have generated a data browser that incorporates functions to allow each of the types of analysis that we highlight below to be conducted for any given source brain region and/or projection target that might be of particular interest (http://neomorph.salk.edu/epiretro).
下面,我们重点介绍这个非常大的数据集的所有可能分析的子集,以突出数据的效用并提供有趣的示例。为了便于对完整数据集的进一步分析,我们生成了一个数据浏览器,其中包含一些功能,允许对可能特别感兴趣的任何给定源大脑区域和/或投射目标 (http://neomorph.salk.edu/epiretro) 进行我们在下面强调的每种类型的分析。
ET- versus IT-projecting neurons
ET 与 IT 投射神经元
In CTX, the most explicit correspondence between projection types and molecular types is observed for neurons that project to ET targets versus IT targets (for a breakdown of ET and IT target regions sampled, see Fig. 1b). To investigate whether such distinctions are shared with neurons from other sources, we explored the genetic distinguishability of neurons projecting to ET versus IT targets across source brain areas. For the cortical source t-distributed stochastic neighbour embedding (t-SNE) plots, the ET-projecting neurons clearly separate into a distinct cluster (layer 5 ET) whereas the IT neurons are found distributed across the annotated IT clusters, as expected (Fig. 2a). ET and IT neurons are also well separated for the projection neurons in the entorhinal cortex (ENT; illustrated in the RHP plot) as well as TH (Fig. 2a), as expected from known projections of glutamatergic TH neurons to cortex versus GABAergic neurons to subcortical targets. ET versus IT neurons show varying levels of separation for the other sources. Generally, comparisons show some degree of separability for each of the source regions, but AUROC scores are higher for cortical sources than for subcortical courses (except TH and AON; Fig. 2b). These observations suggest that ET- versus IT-projecting neurons are not as genetically distinct for other source regions as they are for cortex and TH.
在 CTX 中,对于投射到 ET 目标与 IT 目标的神经元,观察到投射类型和分子类型之间最明确的对应关系(有关采样的 ET 和 IT 目标区域的细分,请参见图 1)。为了研究这种区别是否与其他来源的神经元共享,我们探讨了投射到 ET 的神经元与跨来源大脑区域的 IT 目标的遗传可区分性。对于皮层源 t 分布随机邻域嵌入 (t-SNE) 图,ET 投射神经元显然分离成一个不同的集群(第 5 层 ET),而 IT 神经元则如预期的那样分布在带注释的 IT 集群中(图 D)。ET 和 IT 神经元也很好地分离了内嗅皮层 (ENT;如 RHP 图所示) 以及 TH 中的投射神经元 (图 .2a),正如从谷氨酸能 TH 神经元到皮层的已知投射与 GABA 能神经元到皮层下靶标的预测所预期的那样。ET 与 IT 神经元对其他来源显示出不同程度的分离。一般来说,比较显示每个源区域都有一定程度的可分离性,但皮质源的 AUROC 评分高于皮层下病程(TH 和 AON 除外;无花果。这些观察结果表明,ET 与 IT 投射神经元对于其他源区域的遗传差异不如皮层和 TH 的差异。
图 2:投射到整个大脑中不同目标的神经元的可区分性。

a, Joint t-SNEs of epi-retro-seq, unbiased snmC-seq and scRNA-seq data from ten region groups containing both IT- and ET-projecting neurons from the same source. Only the epi-retro-seq neurons projecting from the same source to ET (blue) and IT (orange) targets are coloured and the other cells are in grey. b, AUROC scores for comparisons between IT versus (vs) ET neurons from each of the 22 source regions. c, AUROC for IT versus ET neurons when the model was trained in one source region (row) and tested on another source region (column). A high AUROC indicates that the epigenetic differences between IT and ET neurons were similar between the training and testing sources. The values on the diagonal of c are the same as the values in b. d, The log2 fold changes of mCH levels in each source at the IT versus ET DMGs. A total of 2,919 DMGs are shown that were identified in at least one of the 22 source regions. The row and column colours represent the region groups in b–d. e, AUROC for comparisons of neurons projecting to each pair of target groups. Each dot represents a comparison in one source region (colour palette as in b). f, AUROC for the comparison of all target pairs from every source region (n = 926) with models using different sets of genes as features. Subsets or all of the 9,906 genes with high coverage in single cells were used. The larger gene sets were downsampled to the same number of genes as the smaller sets for comparison. All comparisons between gene sets are significant (false discovery rates (FDRs) in Supplementary Table 6; two-sided Wilcoxon signed-rank test, Benjamini–Hochberg procedure) except the ones between “neuron projection development” and “neurotransmitter receptor” with 19 genes.
a,epi-retro-seq 的联合 t-SNE,来自十个区域组的无偏 snmC-seq 和 scRNA-seq 数据,包含来自同一来源的 IT 和 ET 投射神经元。只有从同一来源投射到 ET(蓝色)和 IT(橙色)靶点的 epi-retro-seq 神经元是彩色的,其他细胞是灰色的。b,来自 22 个源区域中每个区域的 IT 与 (vs) ET 神经元之间比较的 AUROC 评分。c,当模型在一个源区域(行)进行训练并在另一个源区域(列)进行测试时,IT 神经元与 ET 神经元的 AUROC。高 AUROC 表明 IT 和 ET 神经元之间的表观遗传差异在训练和测试来源之间相似。c 对角线上的值与 b 中的值相同。d,IT 与 ET DMG 中每个来源的 mCH 水平的对数2 倍变化。总共显示了 2,919 个 DMG,这些 DMG 是在 22 个源区域中的至少一个区域中识别的。行和列颜色表示 b-d 中的区域组。e,AUROC 用于比较投射到每对目标组的神经元。每个点代表一个源区域中的比较(调色板如 b 中所示)。f, AUROC,用于将来自每个源区域 (n = 926) 的所有靶标对与使用不同基因集作为特征的模型进行比较。使用单细胞中具有高覆盖率的 9,906 个基因的子集或全部。较大的基因集被下采样到与较小基因集相同的基因数量以进行比较。 除了具有 19 个基因的“神经元投射发育”和“神经递质受体”之间的比较外,所有基因集之间的比较都是显着的(补充表 6 中的错误发现率 (FDR);双侧 Wilcoxon 符号秩检验,Benjamini-Hochberg 程序)。
We next asked whether the epigenetic differences between ET- and IT-projecting neurons are shared across sources or alternatively whether different sources might have distinct molecular signatures that distinguish ET from IT neurons. We trained logistic regression models to distinguish ET- versus IT-projecting neurons in each 1 of the 22 sources, and tested whether each model could accurately separate ET and IT neurons from each of the other sources (Fig. 2c). We observed that the knowledge learned by the models could largely be transferred between isocortical sources and between isocortical and archicortical (ENT and PIR) areas, but not beyond the cortical regions. Other source groups sharing similar ET versus IT differences include MOB and AON, as well as AMY, TH and midbrain reticular nucleus (MRN). To further evaluate these relationships, we identified the differentially methylated genes (DMGs) between ET and IT-projecting cells in each source, which merge into a combined set of 2,919 genes. Consistent with the AUROC results, the mCH levels of these DMGs show similar fold changes across isocortical and archicortical areas, MOB and AON, as well as different parts of TH and MB (Fig. 2d). These observations suggest that the mechanisms that give rise to relationships between projection targets and epigenetics are relatively conserved across cortical areas, across MOB and AON, and across AMY, TH and MRN, but differ between these sets of areas. We further assessed whether neurons projecting to more finely separated groups of targets might be more or less separable. We separated the ET and IT targets into three finer groups (IT: CTX, MOB and CNU; ET: IB, MB and HB) and asked which pairs of target groups are less separable between ET and IT. Most of the target group pairs have better prediction results than ET versus IT, except that the CNU versus IB projecting cells are less distinguishable compared to ET versus IT on the basis of DNA methylomes with linear models (Fig. 2e).
接下来,我们询问了 ET 和 IT 投射神经元之间的表观遗传差异是否在来源之间共享,或者不同的来源是否可能具有不同的分子特征来区分 ET 和 IT 神经元。我们训练了 logistic 回归模型,以区分 22 个来源中每个来源中 1 个中的 ET 和 IT 投射神经元,并测试每个模型是否可以准确地将 ET 和 IT 神经元与其他来源分开(图我们观察到,模型学到的知识可以在很大程度上在等皮层源之间以及等皮层和弓皮层(ENT 和 PIR)区域之间转移,但不能超出皮层区域。其他具有相似 ET 与 IT 差异的来源组包括 MOB 和 AON,以及 AMY、TH 和中脑网状核 (MRN)。为了进一步评估这些关系,我们确定了每个来源中 ET 和 IT 投射细胞之间的差异甲基化基因 (DMG),这些基因合并为一组 2,919 个基因。与 AUROC 结果一致,这些 DMG 的 mCH 水平在等皮层和岛皮质区域、MOB 和 AON 以及 TH 和 MB 的不同部分显示出相似的倍数变化(图 D)。这些观察结果表明,产生投射靶标和表观遗传学之间关系的机制在皮层区域、MOB 和 AON 以及 AMY、TH 和 MRN 之间相对保守,但在这些区域组之间有所不同。我们进一步评估了投射到更精细分离的目标组的神经元是否可能或多或少可分离。我们将 ET 和 IT 目标分为三个更精细的组(IT:CTX、MOB 和 CNU;ET:IB、MB 和 HB),并询问 ET 和 IT 之间哪些目标群体对的可分性较低。 大多数靶组对的预测结果比 ET 与 IT 更好,除了根据线性模型的 DNA 甲基化组,与 ET 与 IT 相比,CNU 与 IB 投射细胞的区分性较差(图 D)。2e)。
To better understand what types of gene are contributing to the predictions of projection targets, we used genes in the following five categories as features to compute AUROC scores: neurotransmitter receptors; neuropeptides and receptors; ion channels; TFs; and neuron projection development (Methods and Supplementary Table 5). As different categories have different numbers of genes, and use of more genes increases prediction performance, we downsampled the larger gene categories into samples including the same numbers of genes as the smaller categories to facilitate comparisons in 5 different groups using from 19 to 666 genes and compared the AUROC scores. We observed that the neuron projection development genes have the strongest target prediction power, followed by neurotransmitter receptors, ion channels, neuropeptides and receptors, TFs, and randomly selected genes (Fig. 2f and Supplementary Table 6). Using all 628 genes in the neuron projection development category achieved an average AUROC of 0.88, which is slightly lower than that using all of the 9,906 genes as features (AUROC 0.91; Fig. 2f), suggesting that additional genes from other Gene Ontology (GO) categories also contribute to the target predictability. The greater predictive power of genes involved in neuron projection development aligns with the idea that similarities and differences between ET- and IT-projecting neurons (from various sources) are tied to developmental mechanisms specifying projection targets.
为了更好地了解哪些类型的基因有助于预测投射目标,我们使用以下五类基因作为特征来计算 AUROC 评分:神经递质受体;神经肽和受体;离子通道;TFs;和神经元投射发育(方法和补充表 5)。由于不同的类别具有不同的基因数量,并且使用更多的基因可以提高预测性能,我们将较大的基因类别下采样为包含与较小类别的相同基因数量的样本,以便于使用 19 到 666 个基因在 5 个不同组中进行比较,并比较 AUROC 评分。我们观察到神经元投射发育基因具有最强的靶点预测能力,其次是神经递质受体、离子通道、神经肽和受体、TFs 和随机选择的基因(图 .2f 和补充表 6)。使用神经元投射发育类别中的所有 628 个基因实现了 0.88 的平均 AUROC,略低于使用所有 9,906 个基因作为特征的 AUROC (AUROC 0.91;无花果。2f),这表明来自其他基因本体 (GO) 类别的其他基因也有助于靶标的可预测性。参与神经元投射发育的基因的更大预测能力与这样一种观点一致,即 ET 和 IT 投射神经元(来自不同来源)之间的相似性和差异性与指定投射目标的发育机制有关。
Hypothalamic projection neurons
下丘脑投射神经元
Analyses of gene expression and DNA methylation patterns have revealed the existence of numerous cell clusters within the HY, indicating a high level of cell-type diversity18,19. Additionally, the HY is composed of many distinct subregions and nuclei, each with unique functions and contributions to innate behaviours such as aggression, mating and feeding20. The HY therefore serves as an excellent use case for our dataset to further examine the relationships between neuronal cell types as defined by their transcriptional and epigenomic signatures, their projection patterns and their spatial organization.
对基因表达和 DNA 甲基化模式的分析揭示了 HY 内存在众多细胞簇,表明细胞类型多样性水平很高18,19。此外,HY 由许多不同的亚区域和核组成,每个亚区域和核都有独特的功能和对先天行为的贡献,例如攻击性、交配和摄食20。因此,HY 是我们数据集的一个很好的用例,可以进一步检查由其转录和表观基因组特征、投影模式和空间组织定义的神经元细胞类型之间的关系。
We profiled hypothalamic neurons that project to ten distinct targets throughout the brain, including prefrontal cortex (PFC), MOB, STR, PAL, AMY, TH, superior colliculus (SC), ventral tegmental area (VTA) and substantia nigra (referred to later as VTA), pons (P) and medulla (MY). By integrating epi-retro-seq data with unbiased snmC-seq and scRNA-seq hypothalamic data, we identified a total of 94 neuronal cell clusters, of which 17 were enriched for the profiled HY projections (Fig. 3a,b). We annotated each co-cluster on the basis of the 302 neuronal cell subclasses identified in scRNA-seq across the whole brain18. Note that this annotation does not give a unique name to each of the finest cluster divisions. Therefore, clusters noted by different identifying cluster numbers (for example, 0 to 94) may share the same cluster name (for example, clusters 0 and 76 were both annotated as STN-PSTN Pitx2 Glut). Each of the projections to the ten targets was enriched in a unique subset of cell clusters (Fig. 3b–d and Supplementary Table 7). For example, HY-to-STR neurons were predominantly enriched in cluster 76, whereas HY-to-AMY neurons were uniquely enriched in cluster 64 (Fig. 3b), indicating distinct cell-type specificity of different HY projection neurons. HY-to-P and HY-to-MY show similar enrichment patterns across clusters, but only HY-to-P neurons were enriched in cluster 76 (Fig. 3b). Similarly, HY-to-PFC and HY-to-MOB neurons were both enriched in cluster 50 and 29, but HY-to-MOB neurons were uniquely enriched in cluster 17 (Fig. 3b). These results indicate that HY neurons projecting to structurally related targets may share some common molecular cell types but also exhibit some level of diversity. These findings underscore the cell-type specificity and diversity of hypothalamic neurons projecting to different targets, shedding light on the potential functional roles of these cell clusters in various physiological and behavioural processes.
我们分析了投射到整个大脑中十个不同目标的下丘脑神经元,包括前额叶皮层 (PFC)、MOB、STR、PAL、AMY、TH、上丘 (SC)、腹侧被盖区 (VTA) 和黑质(后来称为 VTA)、脑桥 (P) 和髓质 (MY)。通过将 epi-retro-seq 数据与无偏倚的 snmC-seq 和 scRNA-seq 下丘脑数据相结合,我们总共鉴定了 94 个神经元细胞簇,其中 17 个富集用于分析的 HY 投影(图 D)。3a,b)。我们根据整个大脑 scRNA-seq 中鉴定的 302 个神经元细胞亚类对每个共簇进行了注释18。请注意,此 annotation 并没有为每个最精细的 cluster divisions 提供唯一的名称。因此,由不同标识群集编号(例如,0 到 94)记录的群集可能共享相同的群集名称(例如,群集 0 和 76 都注释为 STN-PSTN Pitx2 Glut)。对 10 个靶标的每次投射都富集在一个独特的细胞簇子集中(图 D)。3b-d 和补充表 7)。例如,HY-to-STR 神经元主要在簇 76 中富集,而 HY-to-AMY 神经元在簇 64 中独特富集(图 D)。3b),表明不同 HY 投射神经元的不同细胞类型特异性。HY-to-P 和 HY-to-MY 在簇中显示出相似的富集模式,但只有 HY-to-P 神经元在簇 76 中富集(图 7)。同样,HY-to-PFC 和 HY-to-MOB 神经元都在簇 50 和 29 中富集,但 HY-to-MOB 神经元在簇 17 中独特富集(图 1)。这些结果表明,投射到结构相关靶标的 HY 神经元可能共享一些常见的分子细胞类型,但也表现出一定程度的多样性。 这些发现强调了投射到不同靶点的下丘脑神经元的细胞类型特异性和多样性,阐明了这些细胞簇在各种生理和行为过程中的潜在功能作用。
图 3:下丘脑投射神经元的细胞类型、空间位置和基因调控的多样性。

a, Joint t-SNE of epi-retro-seq (n = 1,572), unbiased snmC-seq (n = 11,554) and scRNA-seq (n = 148,840) data for hypothalamic neurons coloured by cell cluster (top) or projection target (bottom, same colour palette as left row colours in Fig. 1b). Seventeen clusters enriched for the profiled projection neurons are outlined. b, The proportion of each of the 10 projections in each of the 17 projection-enriched clusters, z-score normalized across targets. The asterisk denotes enrichment with FDR < 0.01 (one-sided Fisher exact test, Benjamini–Hochberg procedure; Methods). c,d, t-SNE of cells in the 17 projection-enriched clusters coloured by co-cluster. All cells are colored in c, whereas only cells projecting to one target are colored in each of the subplots in d. e,f, Projection-enriched HY clusters mapped to MERFISH data for six coronal slices (e; two biological replicates (R1 and R2) of slices from anterior to posterior (C6, C8 and C10)) and two sagittal slices (f; S1 and S2 from lateral to medial). Examples of clusters with specific spatial locations are labelled in the enlarged insets of each slice. Scale bars, 15 mm. The colour palette for clusters is the same in b–f. g–i, Normalized gene expression (norm expr, g) and gene-body mCH (h) levels of DEGs (n = 1,163) between the 17 projection-enriched clusters, and mCG levels of DEG-associated DMRs (i; n = 148,897), in each cluster. The values are z-score normalized across clusters. The DEGs and cell clusters are arranged in the same orders in g–i. Only the DMRs with the highest anticorrelation with each DEG are shown in i to make the column orders consistent for g–i. Examples of DEGs with GO annotations related to neuronal function and connectivity are labelled on the x axis. j, Examples of TFs whose binding motifs were enriched in hypo-CG-methylated DMRs of each cluster are shown in the bubble plot. The size of each dot represents the enrichment level (area under the curve (AUC)). The colour of the dot indicates the expression (expr.) level of the TF. The clusters are arranged in the same order as in g–i. Cluster 76 was excluded in i,j as the number of cells in the cluster is too small (<30) to be included in the DMR analysis. k, Example triplet combination of TF–CRE–target gene in GRN of HY. Pearson correlation coefficients (PCCs) are shown on edges. The t-SNE plots share the same coordinates as c. Single cells are greyed and shown in the background. Large dots represent clusters, plotted at the mean coordinates of cells in the cluster. The size of each dot is proportional to the number of cells in the cluster. The dots are coloured by Zic1 expression (left), mCG level of an example DMR (middle) and Zic4 expression (right). Colour scales for gene expression are the same as in g, and the colour scale for mCG is the same as in i.
a,由细胞簇(上图)或投影目标(下图)着色的下丘脑神经元的 epi-retro-seq (n = 1,572)、无偏 snmC-seq (n = 11,554) 和 scRNA-seq (n = 148,840) 数据的联合 t-SNE,与图 1 中左行颜色的调色板相同。概述了为分析的投影神经元丰富的 17 个簇。b,17 个投影丰富的集群中每个集群中 10 个投影的比例,跨目标标准化的 z 分数。星号表示 FDR 富集< 0.01(单侧 Fisher 精确检验,Benjamini-Hochberg 程序;方法)。c,d, 17 个富含投影的簇中细胞的 t-SNE,由共簇着色。所有单元格都用 c 着色,而只有投影到一个目标的单元格在 d 的每个子图中着色。e,f,映射到 MERFISH 数据的 6 个冠状切片(e;从前到后切片的两个生物学重复(R1 和 R2)(C6、C8 和 C10))和两个矢状切片(f;S1 和 S2 从外侧到内侧)。具有特定空间位置的聚类示例在每个切片的放大插图中标记。比例尺,15 毫米。群集的调色板在 b-f 中相同。g-i,17 个投射富集簇之间的 DEGs 的标准化基因表达 (norm expr, g) 和基因-体 mCH (h) 水平 (n = 1,163),以及 DEG 相关 DMR 的 mCG 水平 (i;n = 148,897)。这些值是跨集群标准化的 z 分数。 DEG 和单元集群在 g-i 中以相同的顺序排列。i 中仅显示与每个 DEG 反相关程度最高的 DMR,以使 g-i 的列序一致。具有与神经元功能和连接相关的 GO 注释的 DEG 示例在 x 轴上标记。j,气泡图显示了每个簇的结合基序富含低 CG 甲基化 DMR 的 TFs 的实例。每个点的大小表示富集级别(曲线下面积 (AUC))。点的颜色表示 TF 的表达式 (expr.) 级别。聚类的排列顺序与 g-i 中的顺序相同。簇 76 在 i,j 中被排除,因为簇中的细胞数量太少 (<30) 无法包含在 DMR 分析中。k,HY 的 GRN 中 TF-CRE -靶基因的三联体组合示例。Pearson 相关系数 (PCC) 显示在边缘上。t-SNE 图与 c 共享相同的坐标。单个单元格显示为灰色,并显示在背景中。大点表示聚类,绘制在聚类中单元格的平均坐标处。每个点的大小与集群中的单元格数量成正比。点由 Zic1 表达(左)、示例 DMR 的 mCG 水平(中)和 Zic4 表达(右)着色。基因表达的色标与 g 相同,mCG 的色标与 i 相同。
Next we examined the spatial distributions of projection-enriched HY neuron clusters. We carried out MERFISH on both sagittal and coronal brain slices to visualize the spatial location of neurons. By using the gene expression signatures of the projection-enriched clusters, we mapped them to MERFISH cells (Fig. 3e,f and Methods). Notably, most of the 17 projection-enriched clusters were located in different HY subregions, and the spatial distributions of cells from many clusters were distinguished by well-defined boundaries. For instance, clusters 0, 3 and 76 were located in separate ‘stripes’ in the dorsolateral HY, in regions corresponding to zona incerta or subthalamic nucleus (Fig. 3e). With respect to projection targets, some clusters that were enriched for particular projections were relatively confined to specific regions within the HY, whereas other projection-enriched clusters were distributed topographically across the HY. For example, HY-to-TH neurons were enriched in clusters 12, 32 and 3, all of which were located in well-delineated subregions of dorsal HY (Fig. 3b,f). By contrast, the clusters enriched for HB projection neurons were distributed along the anterior to posterior axis of the HY and also occupied locations across the dorsoventral and mediolateral axes (Fig. 3b,f). Overall, our findings underscore the fine-scale spatial organizations of these projection-enriched cell clusters within the HY and the varying degrees of topographical heterogeneity of the locations of projection-defined HY neuronal populations. These observations also highlight the utility of MERFISH data for linking projection cell types to locations that are much smaller than the regions that were dissected.
接下来,我们检查了富含投影的 HY 神经元簇的空间分布。我们在矢状面和冠状状脑切片上都进行了 MERFISH,以可视化神经元的空间位置。通过使用富含投射的簇的基因表达特征,我们将它们定位到 MERFISH 细胞(图 D)。3e,f 和方法)。值得注意的是,17 个富含投影的聚类中的大多数位于不同的 HY 亚区,并且来自许多聚类的单元的空间分布通过明确定义的边界来区分。例如,簇 0、3 和 76 位于背外侧 HY 中的单独“条纹”中,位于对应于不定带或丘脑底核的区域(图 D)。关于投影目标,一些针对特定投影富集的聚类相对局限于 HY 内的特定区域,而其他投影丰富的聚类则分布在 HY 内的地形上。例如,HY-to-TH 神经元在簇 12、32 和 3 中富集,所有这些簇都位于背侧 HY 轮廓分明的亚区(图 D)。3b,f)。相比之下,富含 HB 投射神经元的簇沿 HY 的前轴到后轴分布,并且也占据了背腹轴和内外侧轴的位置(图 D)。3b,f)。总体而言,我们的研究结果强调了 HY 中这些富含投影的细胞簇的精细空间组织,以及投影定义的 HY 神经元群位置的不同程度的地形异质性。这些观察结果还突出了 MERFISH 数据将投影像元类型链接到比剖读区域小得多的位置的实用性。
To gain insight into the molecular characteristics and gene regulation of the projection-enriched clusters, we further used the integrative analysis of epi-retro-seq, snmC-seq and scRNA-seq. We identified 1,163 differentially expressed genes (DEGs) across the 17 clusters in all pairwise comparisons (Fig. 3g). Each cluster has a different set of DEGs, even when there are several clusters enriched for projections to a particular target (note that this contrasts with results for TH below). Notably, many of the DEGs were found to be involved in neuronal function and connectivity, as exemplified by a few highlighted genes (Fig. 3g). mCH levels plotted with an inverted colour map are strikingly similar to the expression levels for the same genes indicating the anticorrelation between methylation and expression of these genes across clusters (Fig. 3h). To investigate the regulation of these DEGs, we identified 148,897 DMRs associated with the DEGs (Methods). The mCG levels of the DEG-associated DMRs exhibited differential methylation patterns consistent with the gene expression and gene-body mCH levels (Fig. 3i). To uncover the regulatory network of these DEGs, we further identified TFs whose binding motifs were enriched in CREs (Fig. 3j), and built a GRN of HY clusters connecting 389 TFs, 46,075 DMRs and 8,184 target genes (Methods). The network captures concordant variation of different data modalities across clusters. For example, Zic1, whose motif is enriched in hypo-CG-methylated DMRs (hypo-DMRs) of clusters 17 and 39, is also expressed at high levels in these clusters. These clusters are located at the anterior ventral part of the HY, and are enriched for neurons projecting to MOB. Another TF-encoding gene expressed at high levels in these clusters, Zic4, was predicted to be a potential target of ZIC1, with 15 DMRs at the flanking region (transcription start site ± 1 megabase) that have ZIC1-binding motifs, and the mCG levels are correlated with the expression of Zic1 and Zic4 (Fig. 3k). The analysis showed some shared sets of TFs between clusters enriched for some projections, such as HY-to-TH. By contrast, more varied sets of TFs were identified between clusters enriched for some other projections, such as HY-to-P or HY-to-MY (Fig. 3j). Additionally, distinct sets of TFs were observed between clusters that were enriched for different projections. Collectively, these findings underscore the existence of diverse GRNs that use distinct TFs and DMRs for different hypothalamic projections. Furthermore, they offer valuable insights into the molecular mechanisms that govern the regulation of projection-enriched cell clusters and their associated genes in the HY.
为了深入了解投射富集簇的分子特征和基因调控,我们进一步使用了 epi-retro-seq 、 snmC-seq 和 scRNA-seq 的综合分析。在所有成对比较中,我们在 17 个簇中鉴定了 1,163 个差异表达基因 (DEG)(图 D)。每个集群都有一组不同的 DEGs,即使有几个集群富集用于对特定目标的预测(请注意,这与下面的 TH 结果形成对比)。值得注意的是,许多 DEGs 被发现参与神经元功能和连接,例如一些突出的基因(图 D)。用倒置色图绘制的 mCH 水平与相同基因的表达水平惊人地相似,这表明甲基化和这些基因跨簇表达之间的反相关(图 D)。为了研究这些 DEGs 的调节,我们确定了 148,897 个与 DEGs 相关的 DMR (方法)。DEG 相关 DMR 的 mCG 水平表现出与基因表达和基因体 mCH 水平一致的差异甲基化模式(图为了揭示这些 DEGs 的调控网络,我们进一步鉴定了其结合基序在 CRE 中富集的 TFs(图 D)。3j),并构建了连接 389 个 TFs、46,075 个 DMR 和 8,184 个靶基因的 HY 簇的 GRN(方法)。该网络捕获了跨集群的不同数据模态的一致变化。例如,Zic1 的基序富含簇 17 和 39 的低 CG 甲基化 DMR (hypo-DMR),在这些簇中也以高水平表达。这些簇位于 HY 的前腹侧部分,并富集了投射到 MOB 的神经元。 在这些簇中高水平表达的另一个 TF 编码基因 Zic4 被预测为 ZIC1 的潜在靶标,在侧翼区域(转录起始位点± 1 兆碱基)有 15 个 DMR,具有 ZIC1 结合基序,并且 mCG 水平与 Zic1 和 Zic4 的表达相关(图 D)。分析显示集群之间一些共享的 TF 集丰富了某些投影,例如 HY-to-TH。相比之下,在为其他一些投影(例如 HY-to-P 或 HY-to-MY)富集的集群之间确定了更多样化的 TF 集(图 D)。此外,在针对不同投影富集的集群之间观察到不同的 TF 集。总的来说,这些发现强调了不同的 GRN 的存在,它们对不同的下丘脑投射使用不同的 TF 和 DMR。此外,它们为控制 HY 中富含投射的细胞簇及其相关基因的调节的分子机制提供了有价值的见解。
In summary, our integrative analysis has revealed the relationships between hypothalamic neurons projecting to ten different targets and their methylation profiles, enrichment in molecular clusters and spatial locations of neurons belonging to those clusters. A previous study linked transcriptomic clusters and their spatial locations within the medial pre-optic area of the HY to specific behaviours21, suggesting that those clusters might mediate their differential contributions to behaviour through differences in their projections. Another study directly linked transcriptomic clusters of neurons and their locations within the ventromedial HY to their projections to the medial pre-optic area or periaqueductal grey by combining retrograde labelling with scRNA-seq and sequential fluorescence in situ hybridization (seqFISH)7. Those experiments revealed projection-enriched clusters, as we have found for a different set of hypothalamic projection targets, but they did not observe clear relationships between transcriptomic clusters, behaviour-specific activation and projections to the periaqueductal grey or medial pre-optic area. We mapped the neurons from ref. 7 to our HY clusters and found that none of the behaviour-enriched clusters or projection-enriched clusters from ref. 7 correspond to any of our projection-enriched clusters in the entire HY (Methods and Extended Data Fig. 7). Our observations across the full spatial extent of HY and a large number of projection targets reveal strong correlations between clusters and projection targets, suggesting that cell types defined by their projections and genetics or epigenetics are also likely to make distinct contributions to hypothalamic function and related behaviours.
总之,我们的综合分析揭示了投射到十个不同靶标的下丘脑神经元与其甲基化谱、分子簇的富集以及属于这些簇的神经元的空间位置之间的关系。之前的一项研究将转录组组簇及其在 HY 内侧视前区域内的空间位置与特定行为联系起来21,表明这些簇可能通过其投射的差异来介导它们对行为的不同贡献。另一项研究通过将逆行标记与 scRNA-seq 和顺序荧光原位杂交 (seqFISH) 相结合,将神经元的转录组簇及其在腹内侧内侧 HY 内的位置与它们向内侧视前区或导水管周围灰色的投射直接联系起来7。正如我们在一组不同的下丘脑投射目标中发现的那样,这些实验揭示了富含投射的簇,但它们没有观察到转录组簇、行为特异性激活和投射到导水管周围灰色或内侧视前区域之间的明确关系。我们将参考文献 7 中的神经元映射到我们的 HY 集群,发现参考文献 7 中的行为丰富的集群或投影丰富的集群都不对应于我们整个 HY 中的任何投影丰富的集群(方法和扩展数据图 .7). 我们对 HY 和大量投影目标的整个空间范围内的观察揭示了集群和投影目标之间的强相关性,这表明由其投影和遗传学或表观遗传学定义的细胞类型也可能对下丘脑功能和相关行为做出不同的贡献。
Thalamic projection neurons
丘脑投射神经元
The TH is a primary hub in sensory and cortical information processing and also projects to subcortical structures. Like HY, TH consists of a large number of nuclei that are organized into many functional groups that are smaller than our dissected regions. The main, central regions of the TH are composed of exclusively excitatory regions (except for a few local GABAergic interneurons in the dorsal lateral geniculate nucleus (LGd)) that are reciprocally connected with cortical areas22. Other more ventral and lateral regions of the TH (such as the ventral lateral geniculate nucleus and reticular thalamic nucleus) contain GABAergic inhibitory neurons that are either reciprocally connected with thalamic excitatory neurons or project to subcortical structures such as the basal ganglia and brainstem22. In contrast to the HY, the TH had a lower degree of cell-type complexity as shown by the smaller number of cell clusters identified through gene expression analysis18. Despite both the TH and HY showing a high level of heterogeneity in their anatomical nuclei and projections, the differences in their cell-type complexity prompted us to investigate whether the relationships between cell types, their projections and their spatial locations in the TH differ from those observed in the HY, as discussed above.
TH 是感觉和皮层信息处理的主要枢纽,也投射到皮层下结构。与 HY 一样,TH 由大量细胞核组成,这些细胞核被组织成许多官能团,这些官能团比我们解剖的区域小。TH 的主要中央区域完全由兴奋区组成(除了背外侧膝状核 (LGd) 中的少数局部 GABA 能中间神经元),这些区域与皮质区域相互连接22。TH 的其他更腹侧和外侧区域(例如腹侧外侧膝状核和网状丘脑核)包含 GABA 能抑制神经元,这些神经元要么与丘脑兴奋性神经元相互连接,要么投射到皮质下结构,例如基底神经节和脑干22。与 HY 相比,TH 的细胞类型复杂性较低,通过基因表达分析鉴定的细胞簇数量较少,这表明18。尽管 TH 和 HY 的解剖核和投射都显示出高度的异质性,但它们细胞类型复杂性的差异促使我们研究细胞类型之间的关系、它们的投射和它们在 TH 中的空间位置是否与在 HY 中观察到的有所不同,如上所述。
We analysed thalamic neurons that project to 12 different targets, including 9 cortical areas (PFC, primary motor cortex (MOp), primary somatosensory cortex (SSp), anterior cingulate cortex (ACA), agranular insular cortex, primary auditory cortex, retrosplenial cortex (RSP), posterior parietal cortex (PTLp) and primary visual cortex (VISp)), SC, VTA and P. To gain a comprehensive understanding of these neurons, we combined epi-retro-seq data with unbiased snmC-seq and scRNA-seq data from the TH. Through this integration, we identified a total of 58 thalamic neuronal cell clusters (Fig. 4a), of which 33 clusters were enriched for epi-retro-seq neurons (Fig. 4b and Supplementary Table 7). It is worth noting that neurons dissected from different anatomical regions within the TH were located in distinct sets of clusters19 (Fig. 4c), as expected from previous descriptions based on analysis of scRNA-seq data8, suggesting that these molecularly defined cell clusters also have a spatial organization.
我们分析了投射到 12 个不同目标的丘脑神经元,包括 9 个皮质区域 (PFC、初级运动皮层 (MOp)、初级体感皮层 (SSp)、前扣带皮层 (ACA)、无颗粒岛叶皮层、初级听觉皮层、脾后皮层 (RSP)、后顶叶皮层 (PTLp) 和初级视觉皮层 (VISp))、SC、VTA 和 P。为了全面了解这些神经元,我们将 epi-retro-seq 数据与来自 TH 的无偏倚 snmC-seq 和 scRNA-seq 数据相结合。通过这种整合,我们总共鉴定了 58 个丘脑神经元细胞簇(图 D)。4a),其中 33 个簇富集了 epi-retro-seq 神经元(图 D)。4b 和补充表 7)。值得注意的是,从 TH 内的不同解剖区域解剖的神经元位于不同的簇集中19(图 1)。4c),正如之前基于 scRNA-seq 数据分析8 的描述所预期的那样,表明这些分子定义的细胞簇也具有空间组织。
图 4:丘脑投射神经元的细胞类型、空间位置和基因调控的多样性。

a, Joint t-SNE of epi-retro-seq (n = 2,606), unbiased snmC-seq (n = 16,943) and scRNA-seq (n = 162,795) data for thalamic neurons coloured by cell cluster. b, The proportion of each of the 12 assayed TH projections in each of the 33 projection-enriched clusters, z-score normalized across targets. The same set of colours is used for labelling clusters in both cluster group 1 (14 clusters) and cluster group 2 (19 clusters). The asterisk denotes enrichment with FDR < 0.01 (one-sided Fisher exact test, Benjamini–Hochberg procedure; Methods). c, TH neurons dissected from two anterior lateral regions (THl-1 and THl-2), two anterior medial regions (THm-1 and THm-2) and three posterior regions (THp-1, THp-2 and THp-3) coloured respectively in the t-SNE. See slices 7–10 in Extended Data Fig. 1 for details. d,e, Projection-enriched TH clusters mapped to MERFISH data for six coronal slices (d; two biological replicates (R1 and R2) of slices from anterior to posterior (C8, C10 and C12)) and two sagittal slices (e; S1 and S2 from lateral to medial). The colours of the clusters in the left (right) column of the insets are the same as for cluster group 1 (2) in b. Examples of clusters with specific spatial locations are labelled in the enlarged insets of each slice. C12R1 and C12R2 are not shown for cluster group 2. Scale bars, 15 mm. f, t-SNE of projection-enriched clusters coloured by co-cluster. The colour palette for clusters in f and the top left of k,l is the same as in b. g–i, Normalized gene expression (norm expr, g) and gene-body mCH (h) levels of DEGs (n = 2,348) between the 33 projection-enriched clusters, and mCG levels of DEG-associated DMRs (i; n = 1,566,402), in each cluster. The values are z-score normalized across clusters. The DEGs and cell clusters are arranged in the same orders in g–i. Only the DMRs with the highest anticorrelation with each DEG are shown in i to make the column orders consistent for g–i. Examples of DEGs with GO annotations related to neuronal function and connectivity are labelled on the x axis. j, Examples of TFs whose binding motifs were enriched in hypo-CG-methylated DMRs of each cluster are shown in the bubble plot. The size of each dot represents the enrichment level (AUC). The colour of the dot indicates the expression level of the TF. The clusters are arranged in the same order as in b,g–i. k,l, Example triplet combinations of TF–CRE–target gene in GRN of TH. PCCs are shown on edges. The coordinates of the t-SNE plots are the same as in f. Top left, cells projecting to SSp (k) or P (l) are highlighted, coloured by co-clusters. In other plots, single cells are greyed and shown as background. Large dots represent clusters, plotted at the mean coordinates of the cells in the cluster. The size of each dot is proportional to the number of cells in the cluster. The dots are coloured by Rora (k) or Pou4f1 (l) expression (top right), mCG level of an example DMR (bottom left) and Sema7a (k) or Irx2 (l) expression (bottom right). The colour scales for gene expression are the same as in g, and the colour scale for mCG is the same as in i.
a,epi-retro-seq (n = 2,606) 的联合 t-SNE,丘脑神经元的无偏倚 snmC-seq (n = 16,943) 和 scRNA-seq (n = 162,795) 数据,由细胞簇着色。b,12 个分析的 TH 投影中每个投影在 33 个富含投影的集群中的每一个的比例,z 分数跨目标标准化。同一组颜色用于标记聚类组 1(14 个聚类)和聚类组 2(19 个聚类)。星号表示 FDR 富集< 0.01(单侧 Fisher 精确检验,Benjamini-Hochberg 程序;方法)。c, 从两个前外侧区域 (THl-1 和 THl-2)、两个前内侧区域 (THm-1 和 THm-2) 和三个后区域 (THp-1 、 THp-2 和 THp-3) 中分离出来的 TH 神经元,分别在 t-SNE 中着色。参见扩展数据图 7-10 中的切片 7-10。1 了解详细信息。d,e,映射到 MERFISH 数据的 6 个冠状切片 (d;从前到后切片的两个生物学重复(R1 和 R2)(C8、C10 和 C12))和两个矢状切片(e;S1 和 S2 从外侧到内侧)。插图左 (右) 列中的聚类颜色与 b 中聚类组 1 (2) 的颜色相同。具有特定空间位置的聚类示例在每个切片的放大插图中标记。集群组 2 的 C12R1 和 C12R2 未显示。比例尺,15 mm. f,由共簇着色的富含投影的簇的 t-SNE。f 和 k,l 左上角的群集的调色板与 b 中的相同。 g-i,33 个投射富集簇之间的 DEG 的标准化基因表达 (norm expr, g) 和基因体 mCH (h) 水平 (n = 2,348),以及 DEG 相关 DMR 的 mCG 水平 (i;n = 1,566,402)。这些值是跨集群标准化的 z 分数。DEG 和单元集群在 g-i 中以相同的顺序排列。i 中仅显示与每个 DEG 反相关程度最高的 DMR,以使 g-i 的列序一致。具有与神经元功能和连接相关的 GO 注释的 DEG 示例在 x 轴上标记。j,气泡图显示了每个簇的结合基序富含低 CG 甲基化 DMR 的 TFs 的实例。每个点的大小表示扩充级别 (AUC)。圆点的颜色表示 TF 的表情级别。这些簇的排列顺序与 b,g–i 中的顺序相同。k,l,TH 的 GRN 中 TF-CRE -靶基因的三联体组合示例。PCC 显示在边缘上。t-SNE 图的坐标与 f 中的坐标相同。左上角,突出显示了投影到 SSp (k) 或 P (l) 的细胞,由共簇着色。在其他绘图中,单个单元格显示为灰色并显示为背景。大点表示聚类,绘制在聚类中单元格的平均坐标处。每个点的大小与集群中的单元格数量成正比。 这些点由 Rora (k) 或 Pou4f1 (l) 表达式(右上)、示例 DMR 的 mCG 水平(左下)和 Sema7a (k) 或 Irx2 (l) 表达式(右下)着色。基因表达的色标与 g 相同,mCG 的色标与 i 相同。
As observed in the HY, each population of thalamic projection neurons exhibited enrichment in distinct subsets of cell clusters, with each cluster showing enrichment for a specific set of projections, sometimes only one (Fig. 4b). Notably, the clusters enriched for TH-to-SC, TH-to-VTA, TH-to-P and TH-to-cortex were mostly mutually exclusive. Regarding cortical projections, TH-to-PTLp and TH-to-VISp neurons exhibited enrichment in a largely overlapping set of clusters, but with varying degrees of enrichment. TH-to-MOp and TH-to-SSp neurons also shared most of their enriched clusters, which differed from those enriched for TH-to-PTLp and TH-to-VISp (Fig. 4b). These results support the notion of a separation of thalamic cell types between the visual and motor pathways in the TH and highlight the heterogeneity of cell types within each pathway. Notably, TH-to-RSP neurons showed no overlap in enriched clusters with any other cortical projections, and were uniquely enriched in clusters 13, 26 and 47 (Fig. 4b). These clusters were annotated by their gene expression patterns as belonging to the anteroventral (AV) nucleus (clusters 13 and 26) and anterodorsal nucleus (cluster 47), which is consistent with TH-to-RSP projections originating from anterior thalamic nuclei23. In summary, TH neurons projecting to cortex versus subcortical targets were enriched in distinct sets of clusters. The enriched cell clusters for cortical projections were further segregated by different thalamic pathways, with several enriched cell clusters observed for each pathway or projection. These findings highlight the cell-type specificity as well as heterogeneity at the level of TH projections.
正如在 HY 中观察到的那样,丘脑投射神经元的每个群体在不同的细胞簇子集中表现出富集,每个簇都显示出一组特定投射的富集,有时只有一个(图 D)。值得注意的是,TH-to-SC、TH-to-VTA、TH-to-P 和 TH-to-cortex 富集的簇大多是互斥的。关于皮层投射,TH-to-PTLp 和 TH-to-VISp 神经元在一组基本重叠的簇中表现出富集,但富集程度不同。TH-to-MOp 和 TH-to-SSp 神经元也共享它们的大部分富集簇,这与 TH-to-PTLp 和 TH-to-VISp 富集的簇不同(图 .这些结果支持 TH 中视觉和运动通路之间丘脑细胞类型分离的概念,并突出了每个通路内细胞类型的异质性。值得注意的是,TH-to-RSP 神经元在丰富的簇中与任何其他皮层投射没有重叠,并且在簇 13、26 和 47 中独特丰富(图这些簇由它们的基因表达模式注释为属于前腹 (AV) 核(簇 13 和 26)和前背核(簇 47),这与源自丘脑前核的 TH-to-RSP 投影一致23。总之,投射到皮层与皮层下靶标的 TH 神经元在不同的簇集中富集。用于皮质投射的富集细胞簇被不同的丘脑途径进一步分离,每个途径或投射都观察到几个富集的细胞簇。这些发现突出了细胞类型特异性以及 TH 投影水平的异质性。
Such cell-type specificity and heterogeneity of TH projection neurons were also reported in transcriptomic analysis of single TH projection neurons (retro-seq)8. Retro-seq neurons of each projection were more similar to epi-retro-seq neurons for the corresponding projections than for any other projections (Extended Data Fig. 8). Small differences between the populations of retrogradely labelled TH neurons in the two datasets are probably due to the use of different injection coordinates for each cortical target (Supplementary Table 8).
在单个 TH 投射神经元的转录组学分析 (retro-seq) 中也报道了 TH 投射神经元的这种细胞类型特异性和异质性8。与任何其他投影相比,每个投影的 retro-seq 神经元与相应投影的 epi-retro-seq 神经元更相似(扩展数据图 .8). 两个数据集中逆行标记的 TH 神经元群之间的微小差异可能是由于每个皮层靶标使用不同的注射坐标(补充表 8)。
As in our approach for the HY, we used the MERFISH data to map the spatial locations of the 33 TH projection-enriched clusters (Fig. 4d,e). Notably, almost all of these clusters exhibited a unique spatial pattern, many of them with distinct boundaries in the distributions of their cells (Fig. 4d,e). These boundaries often corresponded to specific thalamic nuclei, exemplified by clusters 25 and 45 that were enriched for P-projecting neurons and annotated as medial habenula cell types on the basis of their molecular signatures. When mapped to the MERFISH data, cells in these clusters demonstrated a clearly defined spatial location that corresponded to medial habenula (Fig. 4d). This illustrates the high resolution of our data and analysis, enabling the identification of specific medial habenula-to-P projection neurons among all thalamic neurons. Similarly, we were able to accurately map the molecularly annotated anterodorsal cluster 47 and anteroventral cluster 26 that were enriched for the TH-to-RSP projection to their corresponding locations in the dorsal and ventral anterior TH (Fig. 4e). This high resolution of our data also allowed us to investigate the molecular and spatial cellular heterogeneity within a projection. For instance, the visual input from the retina reaches VISp through LGd in TH. When mapped to MERFISH, clusters 34, 5, 4, 1 and 6 that were enriched for TH-to-VISp neurons collectively occupied the location that corresponds to LGd, with each cluster having a unique distribution within LGd (Fig. 4d). These findings underscore the heterogeneity of LGd-to-VISp neurons and provide valuable insights for future in-depth analysis of different types of LGd-to-VISp neurons.
与我们的 HY 方法一样,我们使用 MERFISH 数据来绘制 33 个 TH 投影丰富的集群的空间位置(图 D)。4d,e)。值得注意的是,几乎所有这些星团都表现出独特的空间模式,其中许多星团在其细胞分布中具有不同的边界(图 D)。4d,e)。这些边界通常对应于特定的丘脑核,例如簇 25 和 45,它们富集了 P 投射神经元,并根据其分子特征注释为内侧缰绳细胞类型。当映射到 MERFISH 数据时,这些簇中的细胞表现出与内侧 Habenula 相对应的明确定义的空间位置(图 D)。这说明了我们数据和分析的高分辨率,能够在所有丘脑神经元中识别特定的内侧缰绳到 P 投射神经元。同样,我们能够准确地将分子注释的前背簇 47 和前腹簇 26 映射到它们在背侧和腹侧前部 TH 中的相应位置(图 D)。4e)。我们数据的这种高分辨率还使我们能够研究投影内的分子和空间细胞异质性。例如,来自视网膜的视觉输入通过 TH 中的 LGd 到达 VISp。当映射到 MERFISH 时,针对 TH-to-VISp 神经元富集的簇 34、5、4、1 和 6 共同占据了与 LGd 对应的位置,每个簇在 LGd 中都有独特的分布(图 D)。这些发现强调了 LGd 到 VISp 神经元的异质性,并为未来深入分析不同类型的 LGd 到 VISp 神经元提供了有价值的见解。
Next, we investigated the gene regulation of thalamic neurons in these projection-enriched clusters (Fig. 4f). Joint analysis of scRNA-seq and scmC-seq data for TH identified a total of 2,348 DEGs (Fig. 4g,h) and 1,566,402 associated DMRs (Fig. 4i) across the 33 clusters. As expected, the expression levels of the DEGs (Fig. 4g) were anticorrelated with their mCH levels (Fig. 4h), and their associated DMRs also showed strong correspondence in terms of mCG levels (Fig. 4i). In contrast to HY, TH clusters enriched for the same projections exhibited similar expression patterns of DEGs and methylation patterns of the associated DMRs. Additionally, clusters enriched for the same projection had similar sets of TFs, whereas those enriched for different projections had more distinct sets of TFs (Fig. 4j), implying the existence of projection-specific GRNs. These relationships are in contrast to those observed in HY, where the organization of TF motifs is not closely related to projection targets.
接下来,我们研究了这些富含投影的簇中丘脑神经元的基因调控(图 D)。TH 的 scRNA-seq 和 scmC-seq 数据的联合分析共确定了 2,348 个 DEG(图 D)。4 克,小时)和 1,566,402 个相关 DMR(图 D)。4i) 跨 33 个集群。正如预期的那样,DEGs 的表达水平(图 D)。4g) 与它们的 mCH 水平呈负相关(图4h),它们相关的 DMR 在 mCG 水平方面也显示出很强的对应性(图 D)。与 HY 相比,为相同投影富集的 TH 簇表现出相似的 DEGs 表达模式和相关 DMR 的甲基化模式。此外,为同一投影富集的簇具有相似的 TFs 集,而为不同投影富集的簇具有更不同的 TFs 集(图 D)。4j),这意味着存在特定于投影的 GRN。这些关系与 HY 中观察到的关系形成鲜明对比,在 HY 中,TF 基序的组织与投影目标并不密切相关。
We then constructed a GRN in TH, which consists of 10.9 million TF–DMR–target triplet combinations, involving 469 TFs, 375,279 DMRs and 13,283 target genes. These networks captured regulatory relationships reported in previous studies. For example, RORA has been identified as an essential factor for thalamocortical axon branching24, and transcriptome analysis suggested that Sema7a, another essential regulator of thalamic cortical circuit maturation25, could be a potential target of RORA. In our data, RORA motifs are enriched in many clusters that are enriched for neurons projecting to cortical targets. Similar expression patterns were observed for Rora and Sema7a, as both of these genes are also expressed at high levels in the cortical-projection-enriched clusters. A total of 43 DMRs that potentially mediate this regulation were identified at the flanking region of Sema7a (Fig. 4k and Methods). Our study also suggests new regulatory relationships in TH. POU4F1 has its binding motif enriched in DMRs hypo-methylated in clusters 25 and 45 that make projections to P. The network suggests that genes encoding prepattern TFs IRX1 and IRX2 (ref. 26) are candidate downstream targets of POU4F1, which is also specifically expressed in the same two clusters (Fig. 4l).
然后,我们在 TH 中构建了一个 GRN,它由 1090 万个 TF-DMR-靶三联体组合组成,涉及 469 个 TFs、375,279 个 DMR 和 13,283 个靶基因。这些网络捕获了先前研究中报告的监管关系。例如,RORA 已被确定为丘脑皮质轴突分支的重要因子24,转录组分析表明,丘脑皮质回路成熟的另一种重要调节因子 Sema7a 25 可能是 RORA 的潜在靶标。在我们的数据中,RORA 基序在许多簇中富集,这些簇为投射到皮层靶标的神经元而富集。在 Rora 和 Sema7a 中观察到类似的表达模式,因为这两个基因在富含皮质投射的簇中也以高水平表达。在 Sema7a 的侧翼区域共鉴定出 43 个可能介导这种调节的 DMR(图 D)。4k 和方法)。我们的研究还表明 TH 中存在新的调节关系。POU4F1 的结合基序富含 DMR,在 25 和 45 簇中低甲基化,这些簇投射到 P。该网络表明,编码前模式 TFs IRX1 和 IRX2(参考文献 26)的基因是 POU4F1 的候选下游靶标,POU4F1 也在同一两个簇中特异性表达(图 26)。4l)。
Neurotransmitters in projection neurons
投射神经元中的神经递质
Recent brain-wide single-cell and spatial transcriptomic analyses have revealed remarkable heterogeneity and spatial specificity in neurotransmitter usage among different cell types across the mouse brain18,19. As described above and exemplified in TH and HY, our integrative analysis revealed high levels of cell-type and spatial specificity in neurons with different projections. These findings sparked a further investigation into the neurotransmitter usage of these distinct projection neurons that were in different brain regions and had different cell-type compositions. Insights into the neurotransmitter usage of different projection neurons may shed light on their functional properties and their potential role in behaviour, with broader implications for understanding neural circuits and the mechanisms underlying various brain functions and disorders.
最近的全脑单细胞和空间转录组分析揭示了小鼠大脑中不同细胞类型之间神经递质使用的显著异质性和空间特异性18,19。如上所述,并以 TH 和 HY 为例,我们的综合分析揭示了具有不同投射的神经元中高水平的细胞类型和空间特异性。这些发现引发了对这些位于不同大脑区域且具有不同细胞类型组成的不同投射神经元的神经递质使用情况的进一步研究。深入了解不同投射神经元的神经递质用途可能有助于阐明它们的功能特性及其在行为中的潜在作用,对理解神经回路和各种大脑功能和疾病的潜在机制具有更广泛的意义。
To systematically examine the use of neurotransmitters by different projections, we quantified the levels of expression of nine canonical neurotransmitter transporter genes in each of the projection-enriched clusters within the 12 grouped brain regions described previously (Extended Data Fig. 5). These transporter genes included Slc17a7 (Vglut1), Slc17a6 (Vglut2) and Slc17a8 (Vglut3) for glutamatergic neurons, Slc32a1 (Vgat) for GABAergic neurons, Slc6a2 (Net) for noradrenergic neurons, Slc6a3 (Dat) for dopaminergic neurons, Slc6a4 (Sert) for serotonergic neurons, Slc6a5 (Glyt2) for glycinergic neurons, and Slc18a3 (Vacht) for cholinergic neurons. In addition, we used histidine decarboxylase (Hdc) for histaminergic neurons. Our analysis revealed a diverse range of neurotransmitter usage across the projection-enriched clusters, particularly those in the MB and HB regions. Furthermore, a large proportion of the projection-enriched clusters expressed more than one neurotransmitter transporter gene. These findings indicate that there is a wide variation in neurotransmitter usage across different neural pathways and highlight the heterogeneity within some of these pathways. Below, we discuss a few notable cases in more detail, including projections from the HB regions of P and MY, AMY, and the MB region of VTA.
为了系统地检查不同投射对神经递质的利用,我们量化了前面描述的 12 个分组大脑区域内每个投射丰富的簇中 9 个经典神经递质转运蛋白基因的表达水平(扩展数据图5). 这些转运蛋白基因包括谷氨酸能神经元的 Slc17a7 (Vglut1)、Slc17a6 (Vglut2) 和 Slc17a8 (Vglut3),GABA 能神经元的 Slc32a1 (Vgat),去甲肾上腺素能神经元的 Slc6a2 (Net),多巴胺能神经元的 Slc6a3 (Dat),5-羟色胺能神经元的 Slc6a4 (Sert),甘氨酸能神经元的 Slc6a5 (Glyt2),以及Slc18a3 (Vacht) 用于胆碱能神经元。此外,我们将组氨酸脱羧酶 (Hdc) 用于组胺能神经元。我们的分析揭示了在富含投影的集群中神经递质的使用范围多种多样,尤其是在 MB 和 HB 区域中的神经递质。此外,很大一部分富含投射的簇表达不止一个神经递质转运蛋白基因。这些发现表明,神经递质在不同神经通路中的使用存在很大差异,并突出了其中一些通路内的异质性。下面,我们更详细地讨论了一些值得注意的情况,包括 P 和 MY、AMY 的 HB 区域以及 VTA 的 MB 区域的预测。
Neurotransmitters in HB neurons
HB 神经元中的神经递质
We analysed 11 HB projections, which included projections from P or MY to five different targets—TH, HY, SC, cerebellar nuclei and cerebellar cortex (CBX)—as well as the projection from P to MY. These projections were enriched in 20 cell clusters out of a total of 128 HB clusters. Notably, in both P and MY, neurons projecting to the CBX were the most distinct from other projection neurons (Fig. 5a).
我们分析了 11 个 HB 投影,其中包括从 P 或 MY 到五个不同靶点的投影——TH、HY、SC、小脑核和小脑皮层 (CBX)——以及从 P 到 MY 的投影。这些投影在总共 128 个 HB 簇中的 20 个细胞簇中富集。值得注意的是,在 P 和 MY 中,投射到 CBX 的神经元与其他投射神经元最不同(图 D)。5a)。
图 5:HB、AMY 和 VTA 投射神经元中的神经递质使用情况。

a,b, The proportion of each of the 11 assayed HB projections (a, z-score normalized across targets; the asterisk denotes enrichment with FDR < 0.01 (one-sided Fisher exact test, Benjamini–Hochberg procedure; Methods)) and the expression levels of six neurotransmitter transporter genes, encoding respectively VGLUT1, VGLUT2, VGLUT3, VGAT, SERT and GLYT2 (b), in each of the 20 HB projection-enriched clusters. c, Projection-enriched HB clusters mapped to the MERFISH slice S1. The colour palette for clusters is the same as in a. d, AUC of precision–recall (AUPR) of genes to distinguish the P-to-CBX cluster (0) and P-to-ET clusters (10, 11, 27, 30, 35, 44, 57, 62 and 80) versus the MY-to-CBX cluster (76) and MY-to-ET clusters (5, 7, 10, 11, 17, 57, 66 and 114) with gene-body mCH level in epi-retro-seq data. The genes with AUPR > 0.872 in P and AUPR > 0.647 in MY (>99th quantile) are coloured in red. Five genes selected in both P and MY are labelled. e, mCG levels of hypo-mCG DMRs (n = 22,3839) in P and MY between the P-to-CBX clusters and P-to-ET clusters. f,g, The proportion of each of the 9 assayed AMY projections (f, z-score normalized across targets; the asterisk denotes enrichment with FDR < 0.01 (Fisher exact test, Benjamini–Hochberg procedure; Methods)) and the expression levels of neurotransmitter transporters Slc17a7, Slc17a6 and Slc32a1, encoding respectively VGLUT1, VGLUT2 and VGAT (g), in each of the 16 projection-enriched clusters. h,i, The gene-body mCH levels of tyrosine hydroxylase (Th), Gad2 and Slc17a6 in VTA projection neurons, shown in density plots (h) or scatter plots (i). Colours represent VTA neurons projecting to different targets and the same palette is used in h,i. Note that the x axis in h and both axes in i are plotted as reciprocal mCH values (1/gene-body mCH), so low mCH is plotted to the right and top, indicating higher gene expression. ACA was not included in CTX (see Extended Data Fig. 9).
a,b,11 个测定的 HB 投影中每个投影的比例(a,跨目标标准化的 z 分数;星号表示 FDR < 0.01(单侧 Fisher 精确检验,Benjamini-Hochberg 程序;方法))和 6 个神经递质转运蛋白基因的表达水平,分别编码 VGLUT1、VGLUT2、VGLUT3、VGAT、SERT 和 GLYT2 (b),在 20 个 HB 投射丰富的簇中的每一个中。c,映射到 MERFISH 切片 S1 的投影富集 HB 簇。群集的调色板与 a 中的调色板相同。d,基因的精确召回率 (AUPR) 的 AUC,用于区分 P 到 CBX 簇 (0) 和 P-to-ET 簇 (10、11、27、30、35、44、57、62 和 80) 与 MY-to-CBX 簇 (76) 和 MY-to-ET 簇 (5、7、10、11、17、57、66 和 114) 与 Epi-retro-seq 数据中的基因体 mCH 水平。P 中 AUPR > 0.872 和 MY (>99 分位数) 中 AUPR > 0.647 的基因以红色表示。标记了在 P 和 MY 中选择的 5 个基因。e,P 到 CBX 簇和 P-to-ET 簇之间 P 和 MY 中低 mCG DMR (n = 22,3839) 的 mCG 水平。f,g,9 个分析的 AMY 投影中每个投影的比例(f,跨目标标准化的 z 分数;星号表示 FDR < 0.01(Fisher 精确检验,Benjamini-Hochberg 程序;方法))和神经递质转运蛋白 Slc17a7、Slc17a6 和 Slc32a1 的表达水平,分别编码 VGLUT1、VGLUT2 和 VGAT (g),在 16 个投影丰富的簇中的每一个中。 h,i,VTA 投影神经元中酪氨酸羟化酶 (Th)、Gad2 和 Slc17a6 的基因体 mCH 水平,如密度图 (h) 或散点图 (i) 所示。颜色代表投射到不同目标的 VTA 神经元,在 h,i 中使用相同的调色板。请注意,h 中的 x 轴和 i 中的两个轴都绘制为倒数 mCH 值 (1/基因-体 mCH),因此低 mCH 绘制在右侧和顶部,表明基因表达较高。ACA 未包含在 CTX 中(参见扩展数据图 1)。9).
The 20 projection-enriched clusters showed expression of six neurotransmitter transporter genes (Fig. 5b). Most of these clusters, such as the MY-to-CBX-enriched cluster 76, contain glutamatergic neurons expressing Slc17a6. Notably, Slc17a7 (encoding VGLUT1) and Slc17a6 (encoding VGLUT2) were co-expressed in cluster 0 neurons that were enriched for the P-to-CBX projection. These observations are consistent with those of previous studies that demonstrated the presence of VGLUT1 or VGLUT2 in climbing fibre (MY-to-CBX) terminals and both VGLUT1 and VGLUT2 in cerebellar mossy fibre (P-to-CBX) terminals using synaptic vesicle immunoisolation27. Moreover, different neurotransmitters were used in clusters enriched for the same projections. For instance, clusters 10, 11 and 27 were enriched for P-to-HY projections. Among them, cluster 10 is GABAergic, cluster 11 is glutamatergic, and cluster 27 is serotonergic, showing co-expression of Slc6a4 (encoding SERT) and Slc17a8 (encoding VGLUT3). Furthermore, several of these clusters also exhibited distinctive spatial distributions when mapped to the MERFISH data, such as clusters 0, 76, 10 and 27 (Fig. 5c). Together, these results underscore the extent of molecular, cellular and spatial specificity and diversity within HB projections.
20 个富含投影的簇显示 6 个神经递质转运蛋白基因的表达(图这些簇中的大多数,例如 MY-to-CBX 富集的簇 76,包含表达 Slc17a6 的谷氨酸能神经元。值得注意的是,Slc17a7 (编码 VGLUT1) 和 Slc17a6 (编码 VGLUT2) 在富集用于 P 到 CBX 投影的 0 簇神经元中共表达。这些观察结果与之前的研究一致,这些研究使用突触小泡免疫分离证明攀爬纤维(MY-to-CBX)末端存在 VGLUT1 或 VGLUT2,小脑苔藓纤维(P -to-CBX)末端存在 VGLUT1 和 VGLUT227。此外,在为相同投影富集的簇中使用不同的神经递质。例如,聚类 10 、 11 和 27 为 P 到 HY 投影进行了富集。其中,簇 10 是 GABA 能的,簇 11 是谷氨酸能的,簇 27 是 5-羟色胺能的,显示 Slc6a4 (编码 SERT) 和 Slc17a8 (编码 VGLUT3) 的共表达。此外,当映射到 MERFISH 数据时,其中一些集群也表现出独特的空间分布,例如集群 0、76、10 和 27(图 D)。总之,这些结果强调了 HB 预测中分子、细胞和空间特异性和多样性的程度。
We observed that neurons projecting to CBX from P or MY were distinct from other projections originating from the same regions. To investigate this further, we examined the molecular signatures that could differentiate CBX-projecting neurons from other projection neurons in P or MY. Analysis of gene-body DNA methylation identified genes that could distinguish the P-to-CBX cluster (0) from other projection-associated P clusters, or differentiate the MY-to-CBX cluster (76) from other projection-associated MY clusters (Fig. 5d). Notably, only 5 genes were common between the top 100 genes in the two sets, namely Slit3, Phactr3, Pcbp3, Atp10a and Cdk14 (highlighted in Fig. 5d). Slit3 encodes a repulsive axon guidance molecule28,29, and Phactr3 has been shown to be involved in regulating axonal morphology30,31. The five common genes might mediate functions that are shared between mossy fibres and climbing fibres that are both directed to CBX, whereas the larger numbers of genes that are not shared might be related to distinct functions of MY versus P and/or projections to cerebellar Purkinje cells versus granule cells, respectively. To understand how the DEGs in CBX-projecting neurons are regulated, we identified 223,839 hypo-DMRs in the HB that were associated with CBX-projecting neurons (Fig. 5e). These DMRs were further divided into subsets that were hypo-methylated in either P-to-CBX or MY-to-CBX, and only a limited number were hypo-methylated in both. Collectively, these findings suggest that the molecular mechanisms underlying CBX versus other projections in P and MY are largely distinct, but with some shared features at both the transcriptomic and epigenomic levels.
我们观察到从 P 或 MY 投射到 CBX 的神经元与源自相同区域的其他投射不同。为了进一步研究这一点,我们检查了可以将 CBX 投射神经元与 P 或 MY中的其他投射神经元区分开来的分子特征。基因-体 DNA 甲基化分析确定了可以将 P 到 CBX 簇 (0) 与其他投影相关的 P 簇区分开来,或将 MY-to-CBX 簇 (76) 与其他投影相关的 MY 簇区分开来的基因(图 D)。值得注意的是,在两组中的前 100 个基因中,只有 5 个基因是共同的,即 Slit3、Phactr3、Pcbp3、Atp10a 和 Cdk14(在图 5 中突出显示)。Slit3 编码排斥轴突引导分子28,29,Phactr3 已被证明参与调节轴突形态 30,31。这五个常见基因可能介导苔藓纤维和攀爬纤维之间共享的功能,这些纤维和攀爬纤维都指向 CBX,而大量未共享的基因可能与 MY 与 P 的不同功能和/或投射到小脑浦肯野细胞与颗粒细胞有关,分别。为了了解 CBX 投射神经元中的 DEGs 是如何调节的,我们在 HB 中发现了 223,839 个与 CBX 投射神经元相关的低 DMR(图 D)。这些 DMR 进一步分为在 P 到 CBX 或 MY-到 CBX 中低甲基化的亚群,并且只有有限数量的亚群在两者中都是低甲基化的。 总的来说,这些发现表明,CBX 与 P 和 MY 中的其他投射背后的分子机制在很大程度上是不同的,但在转录组和表观基因组水平上具有一些共同特征。
AMY and MB neurotransmitters
AMY 和 MB 神经递质
We examined projections from the AMY to nine different targets, including the PFC, ENT, HIP, MOB, STR, TH, VTA, P and MY. These projections were enriched in 16 AMY clusters, with distinct sets of clusters enriched for neurons projecting to IT targets versus ET targets (Fig. 5f). The clusters enriched for IT projections were primarily glutamatergic and expressed Slc17a7 and/or Slc17a6 (Fig. 5g). By contrast, the clusters enriched for ET projections were divided between glutamatergic clusters that expressed Slc17a6 and GABAergic clusters (Fig. 5g). Notably, the AMY-to-ENT projection was particularly distinct compared to other IT projections, exhibiting varied usage of vesicular glutamate transporters. Within the clusters enriched for AMY-to-ENT, Slc17a7 was predominantly expressed in cluster 12, Slc17a6 was the predominant transporter in clusters 24, 7 and 1, and clusters 31 and 64 expressed both Slc17a7 and Slc17a6, suggesting a potential diversity in the physiology and function of AMY neurons projecting to the ENT. In summary, our results underscore the heterogeneity in neurotransmitters and their transporter utilization among AMY projection neurons.
我们检查了 AMY 对 9 个不同靶点的预测,包括 PFC、ENT、HIP、MOB、STR、TH、VTA、P 和 MY。这些投影在 16 个 AMY 簇中富集,其中不同的簇集富集针对投射到 IT 目标的神经元而不是 ET 目标(图5f). 为 IT 投射富集的簇主要是谷氨酸能的,并表达 Slc17a7 和/或 Slc17a6 (图 .相比之下,为 ET 投射富集的簇分为表达 Slc17a6 的谷氨酸能簇和 GABA 能簇(图 D)。值得注意的是,与其他 IT 预测相比,AMY 到 ENT 的预测特别明显,表现出囊泡谷氨酸转运蛋白的不同用途。在富含 AMY-to-ENT 的簇中,Slc17a7 主要在簇 12 中表达,Slc17a6 是簇 24、7 和 1 中的主要转运蛋白,簇 31 和 64 同时表达 Slc17a7 和 Slc17a6,表明投射到 ENT 的 AMY 神经元的生理和功能存在潜在多样性。总之,我们的结果强调了 AMY 投射神经元中神经递质及其转运蛋白利用的异质性。
The MB regions containing the VTA and substantia nigra (which we collectively refer to as VTA) exhibit some of the most notable and complex patterns of heterogeneous neurotransmitter usage between different projections. Our study analysed VTA neurons projecting to 16 different targets, including 6 cortical targets (PFC, MOp, SSp, ACA, RSP and PTLp), 6 other IT targets (MOB, ENT, PIR, AMY, STR and PAL) and 4 ET targets (TH, HY, SC and P). By integrating epi-retro-seq and unbiased snmC-seq data, as well as scRNA sequencing of VTA, we can distinguish between cell clusters with various combinations of the expected glutamate, GABA and dopamine transporters known to be expressed by VTA neurons32,33,34,35 (Extended Data Fig. 9a,b).
包含 VTA 和黑质的 MB 区域(我们统称为 VTA)在不同投影之间表现出一些最显着和最复杂的异质神经递质使用模式。我们的研究分析了投射到 16 个不同靶点的 VTA 神经元,包括 6 个皮层靶点(PFC、MOp、SSp、ACA、RSP 和 PTLp)、6 个其他 IT 靶点(MOB、ENT、PIR、AMY、STR 和 PAL)和 4 个 ET 靶点(TH、HY、SC 和 P)。通过整合 epi-retro-seq 和无偏 snmC-seq 数据,以及 VTA 的 scRNA 测序,我们可以区分具有已知由 VTA 神经元表达的预期谷氨酸、GABA 和多巴胺转运蛋白的各种组合的细胞簇32,33,34,35(扩展数据图 3)。9a,b)。
To better examine the relationships between VTA neurons projecting to different targets and their use of neurotransmitters, we analysed the levels of mCH at specific marker genes, including tyrosine hydroxylase (Th) for dopaminergic neurons, Gad2 for GABAergic neurons, and Slc17a6 for glutamatergic neurons because previous studies showed that rodent VTA glutamatergic neurons mainly express Slc17a6 but not Slc17a7 or Slc17a8 (Fig. 5h,i and Extended Data Fig. 9c,d; refs. 36,37). In general, VTA neurons that project to the cortex had lower levels of mCH at Th compared to subcortical projections (except for VTA-to-STR), suggesting a higher expression level of Th (Fig. 5h top; P values = 2.8 × 10−7 (CTX versus MOB), 3.0 × 10−5 (CTX versus PAL), 6.2 × 10−15 (CTX versus ET), two-sided Wilcoxon rank-sum tests). The CTX-projecting neurons also exhibited lower mCH levels at Slc17a6, indicating Slc17a6 expression (Fig. 5h middle). Therefore, these CTX-projecting VTA neurons are probably Th+ and Slc17a6+ and use both dopamine and glutamate (Fig. 5i and Extended Data Fig. 9c). In contrast to VTA-to-CTX neurons, the most prominent populations of VTA-to-STR neurons comprie two groups, Th+Slc17a6− and Th−Slc17a6+, and there is a smaller proportion of neurons that are both Th+ and Slc17a6+ (Fig. 5i and Extended Data Fig. 9). (Note that the use of the “−” designation here indicates a relatively low expression level rather than a complete absence.) On the basis of their mCH levels, the ET-projecting neurons were generally divided into two subgroups: Gad2+ and Slc17a6+ (Fig. 5i). Among the ET-projecting VTA neurons, those projecting to TH and HY were more similar to each other than to those projecting to SC and P (Extended Data Fig. 9b). Notably, some of the SC- and P-projecting neurons were uniquely present in a VTA Gad2+ cluster that were absent in other projections (Extended Data Fig. 9b). Overall, our findings corroborate previous reports of diverse populations of VTA neurons that use single or combined neurotransmitters and highlight intricate patterns of distinct neurotransmitter usage among various projections.
为了更好地检查投射到不同靶标的 VTA 神经元与其对神经递质的使用之间的关系,我们分析了特定标记基因的 mCH 水平,包括多巴胺能神经元的酪氨酸羟化酶 (Th)、GABA 能神经元的 Gad2 和谷氨酸能神经元的 Slc17a6,因为以前的研究表明,啮齿动物 VTA 谷氨酸能神经元主要表达 Slc17a6,而不表达 Slc17a7 或 Slc17a8(图5h,i 和扩展数据图9c,d;裁判。36,37)。一般来说,与皮层下投射相比,投射到皮层的 VTA 神经元在 Th 处的 mCH 水平较低(VTA 到 STR 除外),这表明 Th 的表达水平较高(图 D)。5 小时顶部;P 值 = 2.8 × 10-7(CTX 与 MOB),3.0 × 10-5(CTX 与 PAL),6.2 × 10-15(CTX 与 ET),双侧 Wilcoxon 秩和检验)。CTX 投射神经元在 Slc17a6 处也表现出较低的 mCH 水平,表明 Slc17a6 表达(图5h 中间)。因此,这些投射 CTX 的 VTA 神经元可能是 Th+ 和 Slc17a6+ ,同时使用多巴胺和谷氨酸(图 D)。5i 和扩展数据图与 VTA 到 CTX 神经元相比,VTA 到 STR 神经元中最突出的群体包括两组,Th+Slc17a6− 和 Th−Slc17a6+,并且同时是 Th+ 和 Slc17a6+ 的神经元比例较小(图 D)。5i 和扩展数据图9). (请注意,此处使用 “−” 表示表达水平相对较低,而不是完全不存在。根据它们的 mCH 水平,ET 投射神经元通常分为两个亚组:Gad2+ 和 Slc17a6 +(图在 ET 投射的 VTA 神经元中,投射到 TH 和 HY 的神经元彼此之间的相似性高于投射到 SC 和 P 的神经元(扩展数据图 D)。值得注意的是,一些 SC 和 P 投射神经元独特存在于 VTA Gad2+ 簇中,而其他投影中则不存在(扩展数据图 .总体而言,我们的研究结果证实了先前关于使用单个或组合神经递质的不同 VTA 神经元群的报道,并突出了各种投影中不同神经递质使用的复杂模式。
Summary 总结
We have uploaded and made available data that inform potential users about the relationships between axonal projection status and DNA methylation at single-cell resolution for tens of thousands of neurons corresponding to hundreds of source-to-target combinations. We have provided quantitative measures of the discriminability of source neurons projecting to different targets for nearly 1,000 target-to-target comparisons. We have further demonstrated how these data can be integrated with other single-cell data modalities, including scRNA-seq and MERFISH, to link the spatially resolved cell-type clusters to neural circuits. It is important to note that our experiments were designed to assess the methylation status of neurons projecting to relatively large targets that could be reliably injected and assessed for accuracy during dissections of fresh tissue, and from a large number of source regions that could be readily and reliably dissected. Integration with MERFISH data allowed for more precise anatomical localization of enriched clusters from these sources, but more focused studies using smaller retrograde tracer injections linked to smaller injection locations would be needed to identify possible differences between projection neurons at a finer resolution. More extensive details about the use of these data, their potential limitations and the analytic approaches we have taken can also be found in the Methods. The in-depth analyses provided here for both brain-wide comparisons of ET- versus IT-projecting neurons, and for the full sets of targets assayed for six of the assayed source regions (HY, TH, P, MY, AMY and VTA), exemplify the utility of the much larger dataset for further brain-wide and source- or target-focused analyses.
我们已经上传并提供了数据,告知潜在用户关于对应于数百种源-靶标组合的数万个神经元在单细胞分辨率下轴突投射状态与 DNA 甲基化之间的关系。我们已经为近 1,000 个目标到目标的比较提供了源神经元投射到不同目标的可区分性的定量测量。我们进一步展示了如何将这些数据与其他单细胞数据模式(包括 scRNA-seq 和 MERFISH)集成,以将空间分辨的细胞类型簇与神经回路联系起来。值得注意的是,我们的实验旨在评估投射到相对较大的靶点的神经元的甲基化状态,这些靶点可以可靠地注射,并在新鲜组织解剖过程中评估准确性,以及来自大量可以轻松可靠地解剖的源区域。与 MERFISH 数据的整合允许对来自这些来源的富集簇进行更精确的解剖定位,但需要使用与较小注射位置相关的较小逆行示踪剂注射进行更有针对性的研究,以更精细的分辨率确定投射神经元之间可能存在的差异。有关这些数据的使用、其潜在局限性以及我们采取的分析方法的更广泛详细信息,也可以在方法中找到。这里提供的深入分析既用于 ET 与 IT 投射神经元的全脑比较,也用于六个检测源区域(HY、TH、P、MY、AMY 和 VTA)的全套靶标,体现了更大的数据集用于进一步的全脑和源或靶标分析的效用。
The observations and analysis presented here, both across the whole brain and for selected regions, provide new knowledge about the relationships between projection cell types and their epigenetics and gene expression. Overall, our data and analyses suggest that, as a general rule, the targets of projection neurons in any part of the brain can be predicted at levels above chance on the basis of knowledge of DNA methylation (for example, Extended Data Fig. 3). However, there is considerable diversity in the level of correlation between projection targets and methylation status. This diversity arises from differences in both source regions and targets. For example, cortical neurons projecting to ET versus IT targets can be readily identified for neurons from nearly any cortical area, and knowledge about correlations observed in one cortical area can be used to make predictions for another cortical area. Thalamic neurons projecting to ET versus IT targets can also be readily predicted, but knowledge from differential methylation of cortical neurons cannot be used to accurately predict the projection status of thalamic neurons. In contrast to cortical and thalamic sources, projections to ET versus IT targets cannot be predicted as reliably for neurons in some other sources such as PAL (Fig. 2b). It is also clear from our in-depth analyses of subsets of these data, including neurons in the HY and TH, that relationships between source locations, projection targets and methylation status of single neurons are complex. Although there are better than chance correlations between gene methylation and projection targets for neurons in all of the sources that we sampled, it is likely that the degree of correlation is shaped by a range of developmental events that affect both gene methylation and projection status through mechanisms that can work both independently and in concert. Future studies will be needed to better understand such developmental mechanisms in the context of the whole brain and to study which mechanisms are at work for each brain area and projection target.
这里介绍的在整个大脑和选定区域的观察和分析,为投射细胞类型与其表观遗传学和基因表达之间的关系提供了新的知识。总体而言,我们的数据和分析表明,作为一般规则,大脑任何部分的投射神经元的目标都可以根据 DNA 甲基化知识在高于偶然性的水平上预测(例如,扩展数据图 1 月。然而,投射靶标和甲基化状态之间的相关性水平存在相当大的差异。这种多样性源于来源区域和靶标的差异。例如,对于几乎任何皮层区域的神经元,可以很容易地识别投射到 ET 与 IT 目标的皮层神经元,并且有关在一个皮层区域中观察到的相关性的知识可用于预测另一个皮层区域。投射到 ET 与 IT 目标的丘脑神经元也很容易预测,但皮质神经元差异甲基化的信息不能用于准确预测丘脑神经元的投射状态。与皮质和丘脑来源相比,对于其他一些来源(如 PAL)中的神经元,无法可靠地预测 ET 与 IT 目标的投射(图 D)。从我们对这些数据子集(包括 HY 和 TH 中的神经元)的深入分析中也可以清楚地看出,源位置、投射靶标和单个神经元的甲基化状态之间的关系很复杂。 尽管在我们采样的所有来源中,基因甲基化和神经元的投射靶标之间的相关性优于偶然性,但相关性的程度很可能是由一系列发育事件决定的,这些事件通过既独立又协同工作的机制影响基因甲基化和投射状态。需要进一步的研究来更好地了解整个大脑背景下的这种发展机制,并研究哪些机制对每个大脑区域和投射目标起作用。
Methods 方法
Experimental animals 实验动物
As described previously2, all experimental procedures using live animals were approved by the Salk Institute Animal Care and Use Committee. The knock-in mouse line, R26R-CAG-loxp-stop-loxp-Sun1-sfGFP-Myc (INTACT) used in epi-retro-seq2 was maintained on a C57BL/6J background. Adult male and female INTACT mice were used for the retrograde labelling experiments. Animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility at the Salk Institute. Lighting was controlled on a 12 h light/12 h dark cycle. Temperature was monitored and adjusted in accordance with Guide for the Care and Use of Laboratory Animals. Humidity was not controlled but monitored. As all air coming in is 100% fresh air (not recirculated), humidity in the animal facilities is approximately the same as the outside ambient air. San Diego averages 40–60% humidity year-round. Animals were 35–54 days old at the time of surgery for viral vector injections, were killed 13–17 days later, and were 50–70 days old on the day of dissection. C57BL/6J ‘wild-type’ mice aged 56–63 days were used for MERFISH experiments.
如前所述2,所有使用活体动物的实验程序均已获得索尔克研究所动物护理和使用委员会的批准。epi-retro-seq2 中使用的敲入小鼠系 R26R-CAG-loxp-stop-loxp-Sun1-sfGFP-Myc (INTACT) 维持在 C57BL/6J 背景上。成年雄性和雌性 INTACT 小鼠用于逆行标记实验。动物被安置在索尔克研究所的实验动物护理评估和认证协会认可的设施中。在 12 小时光照/12 小时黑暗周期中控制照明。根据实验动物护理和使用指南监测和调整体温。湿度不受控制,但受到监控。由于所有进入的空气都是 100% 新鲜空气(未再循环),因此动物设施内的湿度与外部环境空气大致相同。圣地亚哥全年平均湿度为 40-60%。动物在病毒载体注射手术时为 35-54 天大,13-17 天后被杀死,解剖当天为 50-70 天大。56-63 天的 C57BL/6J '野生型' 小鼠用于 MERFISH 实验。
Surgical procedures for viral vector injections
病毒载体注射的外科手术
As described previously2, to label neurons projecting to regions of interest, injections of rAAV-retro-Cre (produced by Salk Vector Core or Vigene, 2 × 1012 to 1 × 1013 viral genomes per millilitre, produced with capsid from Addgene plasmid No. 81070 packaging pAAV-EF1a-Cre from Addgene plasmid No. 55636) were made into both hemispheres of the INTACT mice. In summary, animals were anaesthetized with either ketamine–xylazine or isoflurane and placed in a stereotaxic frame. Pressure injections of 0.05 to 0.4 μl of AAV per injection site were made using glass micropipettes (tip diameters about 10–30 μm) targeted to stereotaxic coordinates corresponding to MOp, SSp, ACA, AUDp, RSP, PTLp, VISp, HPF, MOB, STR, PAL, TH, SC, VTA + substantia nigra, P, MY and CBX. To precisely target PFC, agranular insular cortex, ENT, PIR, AMY, HY and CBN, AAV was injected using iontophoresis to ensure confined viral infection. Iontophoretic injections (+5 µA, 7 s on/7 s off cycles for 5–10 min) were made with glass pipettes with a tip diameter of about 10 μm. For most of the desired target areas, injections were made at different depths, and/or at different anterior–posterior or medial–lateral coordinates to label neurons throughout the target area. More detailed injection coordinates and conditions are listed in Supplementary Table 1. At least two male and two female mice were injected for each desired target. No sample size calculation was carried out. We empirically determined to use two mice of the same sex for each injection to achieve minimum reproducibility. Animals used for injections into each brain area were selected at random.
如前所述2,为了标记投射到感兴趣区域的神经元,将 rAAV-retro-Cre(由 Salk Vector Core 或 Vigene 产生,每毫升 2 × 1012 至 ×1013 个病毒基因组,用 Addgene 质粒 81070 号的衣壳产生,包装来自 Addgene 质粒 55636 的 pAAV-EF1a-Cre)被制备到 INTACT 小鼠的两个半球中。总之,用氯胺酮-甲苯噻嗪或异氟醚麻醉动物,并将其置于立体定位框架中。使用玻璃微量移液器(尖端直径约 10-30 μm)对对应于 MOp、SSp、ACA、AUDp、RSP、PTLp、VISp、HPF、MOB、STR、PAL、TH、SC、VTA + 黑质、P、MY 和 CBX 的立体定位坐标,每个注射部位进行 0.05 至 0.4 μl AAV 的压力注射。为了精确靶向 PFC、无颗粒岛叶皮层、ENT、PIR、AMY、HY 和 CBN,使用离子电渗疗法注射 AAV 以确保局限性病毒感染。离子电渗注射(+5 μA,7 s 开/7 s 关循环,5-10 分钟)使用吸头直径约为 10 μm 的玻璃移液器进行。对于大多数所需的目标区域,在不同深度和/或不同的前-后或内侧-外侧坐标进行注射,以标记整个目标区域的神经元。更详细的注射坐标和条件列于补充表 1 中。针对每个所需靶标至少注射两只雄性和两只雌性小鼠。未进行样本量计算。我们凭经验确定每次注射使用两只同性别的小鼠,以实现最小的可重复性。用于注射到每个大脑区域的动物是随机选择的。
Brain dissection 脑解剖
Brain dissections were carried out as described previously2. In summary, approximately 2 weeks after the AAV-retro-Cre injection, brains were extracted from the 50- to 70-day-old INTACT mice, immediately submerged in ice-cold slicing buffer (2.5 mM KCl, 0.5 mM CaCl2, 7 mM MgCl2, 1.25 mM NaH2PO4, 110 mM sucrose, 10 mM glucose and 25 mM NaHCO3) that was bubbled with carbogen, and sliced into 0.6-mm coronal sections starting from the frontal pole. From each AAV-retro-Cre-injected brain, the slices were kept in the ice-cold dissection buffer from which selected brain regions (Fig. 1b) were manually dissected under a fluorescence dissecting microscope (Olympus SZX16), following the Allen Mouse Common Coordinate Framework, Reference Atlas, Version 3 (2015; Extended Data Fig. 1). The dissected brain tissues were transferred to prelabelled microcentrifuge tubes, immediately frozen in dry ice, and subsequently stored at −80° C. During the dissection, the injection site was visually inspected to verify the accuracy of the injection. Only brains with accurate injections were dissected for further analysis. Olympus cellSens dimension 1.8 was used for image acquisition.
如前所述进行脑清扫2.总之,在 AAV-retro-Cre 注射后约 2 周,从 50 至 70 日龄的 INTACT 小鼠中提取大脑,立即浸没在冰冷的切片缓冲液(2.5 mM KCl、0.5 mM CaCl2、7 mM MgCl2、1.25 mM NaH2PO4、110 mM 蔗糖、10 mM 葡萄糖和 25 mM NaHCO3)中,用碳素鼓泡, 并从额极开始切成 0.6 毫米的冠状切片。从每个 AAV 逆向 Cre 注射的大脑中,切片保存在冰冷的解剖缓冲液中,从中选择的大脑区域(图 D)。1b) 在荧光解剖显微镜 (Olympus SZX16) 下手动解剖,遵循 Allen Mouse Common Coordinate Framework, Reference Atlas, Version 3 (2015;扩展数据 图将解剖的脑组织转移到预先标记的微量离心管中,立即在干冰中冷冻,随后储存在 -80°C。在解剖过程中,目视检查注射部位以验证注射的准确性。仅解剖注射准确后的大脑以进行进一步分析。使用 Olympus cellSens 尺寸 1.8 进行图像采集。
Nucleus preparation and single-nucleus isolation
细胞核制备和单细胞核分离
Nucleus preparation and isolation were carried out as described previously2. In summary, for each dissected brain region, samples from two male and two female mice were pooled separately as biological replicates for nucleus preparation. Nuclei were prepared using a modified protocol as reported38 and described2 previously. Nucleus suspensions were then incubated with GFP antibody, Alexa Fluor 488 (Invitrogen, A-21311, 1:500 dilution) and anti-NeuN antibody (EMD Millipore MAB377) conjugated with Alexa Fluor 647 (Invitrogen A20173; 1:300 dilution). GFP+NeuN+ single nuclei were isolated using FANS on a BD Influx sorter or a BD Aria Fusion cell sorter with a 100-μm nozzle, and sorted into 384-well plates with digestion buffer for snmC-seq. BD Influx Software v1.2.0.142 was used to select cell populations. The collected plates were incubated at 50° C for 20 min and then stored at −20° C. FACS parameters were adjusted for each experiment on the basis of the density of labelled neurons such that a higher proportion of all labelled neurons would be recovered from regions with sparser labelling.
细胞核制备和分离如前所述进行2.总之,对于每个解剖的大脑区域,将来自两只雄性和两只雌性小鼠的样本分别合并作为生物重复进行细胞核制备。使用修改后的方案制备细胞核,如之前报道的38 和描述2 所示。然后将细胞核悬液与 GFP 抗体、Alexa Fluor 488(Invitrogen,A-21311,1:500 稀释)和抗 NeuN 抗体(EMD Millipore MAB377)与 Alexa Fluor 647(Invitrogen A20173;1:300 稀释)偶联一起孵育。在 BD Influx 分选仪或带有 100 μm 喷嘴的 BD Aria Fusion 细胞分选仪上使用 FANS 分离 GFP+NeuN+ 单细胞核,并使用 snmC-seq 消化缓冲液分选到 384 孔板中。使用 BD Influx 软件 v1.2.0.142 选择细胞群。将收集的板在 50°C 下孵育 20 分钟,然后储存在 -20°C。根据标记神经元的密度为每个实验调整 FACS 参数,以便从标记稀疏的区域恢复更高比例的所有标记神经元。
snmC-seq library preparation
snmC-seq 文库制备
The bisulfite conversion and library preparation were carried out following the detailed snmC-seq protocol as previously described15. In brief, DNA samples from single nuclei were barcoded with random primers after the bisulfite conversion, pooled through two rounds of cleaning up with SPRI beads, and then added with adapters and PCR amplified to generate the libraries. Libraries were then pooled, cleaned up with SPRI beads, normalized and sequenced on Illumina Novaseq 6000 using the S4 flow cell 2 × 150 base-pair mode. Freedom EVOware v2.7 was used for library preparation, and Illumina MiSeq control software v3.1.0.13 and NovaSeq 6000 control software v.1.6.0 and Real-Time Analysis v3.4.4 were used for sequencing. Technicians doing nucleus preparations and snmC-seq analyses were blind to the injection sites used for each sample.
亚硫酸氢盐转化和文库制备按照前面描述的详细 snmC-seq 方案进行15。简而言之,亚硫酸氢盐转化后,用随机引物对来自单个细胞核的 DNA 样品进行条形码编码,用 SPRI 珠子通过两轮纯化进行合并,然后加入接头和 PCR 扩增以生成文库。然后混合文库,用 SPRI 微珠纯化,在 Illumina Novaseq 6000 上使用 S4 流通池 2 × 150 碱基对模式进行归一化和测序。使用 Freedom EVOware v2.7 进行文库制备,使用 Illumina MiSeq 控制软件 v3.1.0.13 和 NovaSeq 6000 控制软件 v.1.6.0 和 Real-Time Analysis v3.4.4 进行测序。进行细胞核制备和 snmC-seq 分析的技术人员对每个样品使用的注射部位不知情。
Mapping and preprocessing
映射和预处理
Epi-retro-seq data were mapped to the mm10 genome as described in our previous study39. The whole genome was parsed into 100 kb non-overlapping genomic bins (Chr. 1:0–100,000; Chr. 1: 100,000–200,000; and so on) using bedtools make-window, and for each single cell, we counted the methylated and total basecalls for all 100-kb bins using ALLCools generate-dataset. We also carried out the same counting on all gene bodies expanded 2 kb in both directions. The data are saved in Zarr format to allow chunk loading and on-disk computing40. To avoid the methylation differences being driven by the active and inactive X chromosomes, we used only the autosomal bins and genes in our analyses. The cell-by-bin and cell-by-gene posterior methylation levels were computed as previously described39, which is the input for all downstream analyses.
正如我们之前的研究39 中所述,Epi-retro-seq 数据被映射到 mm10 基因组。使用 bedtools make-window 将整个基因组解析为 100 kb 的非重叠基因组 bins(1:0–100,000;1hr. 1:100,000–200,000;依此类推),对于每个细胞,我们使用 ALLCools generate-dataset 计算所有 100 kb bins 的甲基化和总碱基调用。我们还对两个方向扩增 2 kb 的所有基因体进行了相同的计数。数据以 Zarr 格式保存,以允许块加载和磁盘计算40.为了避免由活性和非活性 X 染色体驱动的甲基化差异,我们在分析中仅使用了常染色体 bin 和基因。如前所述计算 cell-by-bin 和 cell-by-gene 后验甲基化水平39,这是所有下游分析的输入。
Quality control 质量管理
In quality control (QC) step 1, the cells included in the analysis are required to have a median mCCC level of the experiment < 0.025; 500,000 < nonclonal reads < 10,000,000; and mCCC level < 0.05. In total, 56,843 cells from 703 experiments satisfied these requirements (Extended Data Fig. 2a,b).
在质量控制 (QC) 步骤 1 中,分析中包含的细胞要求实验 < 的中位 mCCC 水平为 0.025;500,000 < 非克隆读数 < 10,000,000;和 mCCC 水平 < 0.05。总共来自 703 个实验的 56,843 个细胞满足这些要求(扩展数据图 D)。2a,b)。
In QC step 2, the potential doublets were removed as described in the next section, and 48,032 cells remained in the dataset (Extended Data Fig. 2c,d). The cell-type information and dissection information for these cells were used in our analysis, but further filters were applied to exclude non-neuronal cells as well as neurons whose projection targets are not confidently assigned.
在 QC 步骤 2 中,如下一节所述去除了潜在的双峰,数据集中仍保留了 48,032 个细胞(扩展数据图 .2c,d)。我们的分析中使用了这些细胞的细胞类型信息和解剖信息,但应用了进一步的过滤器来排除非神经元细胞以及投影目标没有确定分配的神经元。
In QC step 3, the experiments with fewer than 20 neurons were excluded to ensure the statistical power of projection analysis, resulting in 39,461 cells from 519 experiments left. The non-neuronal cells were also removed from the dataset, after which 34,643 neurons remained. The cell-type classification method is described in the next section.
在 QC 步骤 3 中,排除了少于 20 个神经元的实验,以确保投影分析的统计功效,结果从 519 个实验中留下了 39,461 个细胞。非神经元细胞也从数据集中删除,之后还剩下 34,643 个神经元。下一节将介绍像元类型分类方法。
In QC step 4, the cortical cells from 286 experiments were further filtered to exclude the experiments with a high proportion of neurons of the cell types known not to project to the intended injection site (off-target clusters), using the same method as in our previous study2. Specifically, for each FANS run, we counted the number of neurons that were observed in known on-target cell types () and off-target cell types (). Assuming that the proportions of contaminated cells in each subclass would be similar to those of a sample without projection-type enrichment, we compared the observed counts to the counts from unbiased snmC-seq data ( and ) collected from the corresponding dissections in Extended Data Fig. 1. The fold enrichment was computed as . A one-sided exact binomial test of goodness-of-fit was used to determine whether the enrichment of on-target cells was significant. The P value was computed as , in which , where ~ represents distributed as, and . For each ET target, we considered ET as on-target subclasses and IT+inhibitory neurons as off-target. The thresholds for fold enrichment and FDR (Benjamini–Hochberg procedure) were 8 and 0.001. For IT targets, we considered IT as on-target subclasses and layer 6 corticothalamic+inhibitory neurons as off-target. The thresholds for fold enrichment and FDR (Benjamini–Hochberg procedure) were 3 and 0.001. This eliminated 32 out of 286 sorting cases (Extended Data Fig. 2e).
在 QC 步骤 4 中,使用与我们之前研究 2 相同的方法,进一步过滤来自 286 个实验的皮质细胞,以排除已知不投射到预期注射部位(脱靶簇)的高比例细胞类型的神经元的实验2。具体来说,对于每次 FANS 运行,我们计算了在已知的靶向细胞类型 ( ) 和脱靶细胞类型 ( ) 中观察到的神经元数量。 假设每个亚类中受污染细胞的比例与没有投影型富集的样本的比例相似,我们将观察到的计数与从扩展数据中的相应解剖中收集的无偏性 snmC-seq 数据 ( 和 ) 的计数进行了比较。1. 折叠富集计算为 。使用拟合优度的单侧精确二项式检验来确定靶向细胞的富集是否显著。P 值计算为 ,其中 ,其中 ,其中 ~ 表示分布为 和 。对于每个 ET 靶标,我们将 ET 视为靶向亚类,将 IT + 抑制性神经元视为脱靶。折叠富集和 FDR(Benjamini-Hochberg 手术)的阈值分别为 8 和 0.001。对于 IT 靶点,我们将 IT 视为靶向亚类,将第 6 层皮质丘脑 + 抑制性神经元视为脱靶。折叠富集和 FDR(Benjamini-Hochberg 手术)的阈值分别为 3 和 0.001。这消除了 286 个分拣案例中的 32 个(扩展数据图 .2e)。
The rationale of QC step 4 is to remove potential contamination in the dataset that might have resulted from inaccurate gating of GFP+NeuN+ cells and AAV-retro-Cre injection pipettes that passed through overlying source brain regions and directly labelled neurons at those sources rather than being taken up retrogradely from the intended target. Inaccurate gating of GFP+NeuN+ cells could be more common in the experiments of some weak projections, in which very few neurons were retrogradely labelled, resulting in small proportions of cells passing FANS gating criteria and subsequent inclusion of high proportions of cells accepted from the edges of FANS gates. Inaccurate labelling could be more common when targeting a deep structure in the brain (for example, TH or HY) and collecting cells from the superficial structures directly above the target (for example, cortex). Note that QC step 4 was carried out only for experiments on isocortical neurons, given that the on-target and off-target clusters were relatively clear in these areas. For subcortical projections, comprehensive prior knowledge of molecular cell types associated with projection is usually lacking, which makes the estimation of contamination using this method more challenging. The projections profiled in the subcortical structures are usually strong and do not involve overlaying of sources and targets, which would potentially lead to a lower noise level in those data. Nevertheless, it is worth noting that even after these QC steps, there are still expected to be some contaminated cells remaining in the dataset.
QC 步骤 4 的基本原理是去除数据集中的潜在污染,这些污染可能是由于 GFP + NeuN + 细胞和 AAV-retro-Cre 注射移液管的门控不准确而导致的,这些移液管穿过上覆的源脑区域并直接标记这些源的神经元,而不是从预期目标逆行摄取。在一些弱投影的实验中,GFP + NeuN + 细胞的不准确设门可能更为常见,其中很少有神经元被逆行标记,导致一小部分细胞通过 FANS 门控标准,随后包含高比例的细胞来自 FANS 门的边缘。当靶向大脑中的深层结构(例如 TH 或 HY)并从靶标正上方的浅表结构(例如皮层)收集细胞时,不准确的标记可能更常见。请注意,QC 步骤 4 仅用于同等皮质神经元的实验,因为在这些区域靶向和非靶向簇相对清晰。对于皮层下投射,通常缺乏与投射相关的分子细胞类型的全面先验知识,这使得使用这种方法估计污染更具挑战性。在皮层下结构中分析的投影通常很强,并且不涉及源和目标的叠加,这可能会导致这些数据中的噪声水平降低。然而,值得注意的是,即使在这些 QC 步骤之后,预计数据集中仍会残留一些受污染的细胞。
After all of the QC steps, 33,304 neurons from 487 experiments were used for analyses related to projection targets.
在所有 QC 步骤之后,来自 487 个实验的 33,304 个神经元用于与投影靶标相关的分析。
Transfer of cell labels from one dataset to another with weighted k-nearest neighbours
将单元格标签从一个数据集传输到另一个具有加权 k 最近邻的数据集
This method is similar to the label transfer method in Seurat v3 (ref. 41), and implemented in our ALLCools python package. This is used in many analyses throughout the manuscript, including epi-retro-seq cell classification and doublet removal, and mapping of MERFISH cells and retro-seq cells into major dissection regions or RNA and mC co-clusters. The original Seurat method identified anchors between two datasets, and used the 100 nearest anchors for each cell in the unlabelled dataset to average the information from the labelled dataset. As the 100 anchors usually include cells from other clusters, especially for a cell in an underrepresented cluster, this method makes the label transfer of small clusters quite noisy. Instead of using the anchors between datasets to transfer the labels, we used the anchors only to integrate the datasets together, and directly find the neighbouring cells of the unlabelled dataset in the labelled dataset on the integrated space. As the larger dataset usually has more cells than the number of anchors, this method reduced the noise in the small clusters.
此方法类似于 Seurat v3(参考文献 41)中的标签传输方法,并在我们的 ALLCools python 包中实现。这用于整个手稿中的许多分析,包括 epi-retro-seq 细胞分类和双峰去除,以及将 MERFISH 细胞和 retro-seq 细胞定位为主要解剖区域或 RNA 和 mC 共簇。最初的 Seurat 方法识别两个数据集之间的锚点,并对未标记数据集中每个单元格使用 100 个最近的锚点来平均标记数据集中的信息。由于 100 个锚点通常包括来自其他聚类的单元格,尤其是对于代表性不足的聚类中的单元格,这种方法使小聚类的标签传输非常嘈杂。我们没有使用数据集之间的锚点来传递标签,而是只使用锚点将数据集整合在一起,并在积分空间上的标记数据集中直接找到未标记数据集的相邻单元格。由于较大的数据集通常具有比锚点数量更多的单元格,因此这种方法减少了小集群中的噪声。
Assume we have two datasets in a co-embedding space, A with labels and B without labels. For each cell in B as a query cell, we first find its k-nearest neighbours in A with Euclidean distance, and denote its distances to the neighbours as a k-dimensional vector d. d is then transformed to w as the weights for averaging the information from the neighbours through the following steps that are the same as in Seurat: ; ; . After the transformation, the closer neighbours have higher weights, and the weights of all neighbours sum up to 1. To transfer a categorical label from A to B, we used one-hot encoding to represent the label and the label vectors corresponding to the k neighbours in A of the query cell (k-by-#categories, denoted as ) were averaged with the weights w. The resulting vector represents the probability of the query cell belonging to each category. The category with the maximum probability is used as the final assignment.
假设我们在共嵌入空间中有两个数据集,A 有标签,B 没有标签。对于 B 中的每个单元格作为查询单元格,我们首先在 A 中找到它在 A 中具有欧几里得距离的 k 个最近的邻居,并将它与邻居的距离表示为 k 维向量 d。然后将 d 转换为 w 作为权重,通过以下步骤对来自邻居的信息进行平均,这与修拉中相同: ; ; 。变换后,距离较近的邻居具有较高的权重,所有邻居的权重之和为 1。为了将分类标签从 A 转移到 B,我们使用 one-hot 编码来表示标签,并将对应于查询单元格 A 中的 k 个邻居(k×#categories,表示为 )的标签向量与权重 w 取平均值。结果向量 表示查询单元格属于每个类别的概率。具有最大概率的类别将用作最终作业。
Cell classification and doublet removal
细胞分类和双峰去除
As described in our companion manuscript, the cell clustering of the unbiased dataset was carried out iteratively at four levels (L1–L4), which assigned the cells into 61 (L1), 411 (L2), 1,346 (L3) and 2,573 (L4) clusters, respectively. At each level, the highly variable 100-kb bins were selected, and principal component analysis (PCA) was used for dimension reduction. The significant principal components (PCs) from mCH and mCG were combined to carry out consensus clustering.
正如我们的配套手稿中所述,无偏数据集的细胞聚类在四个级别 (L1-L4) 迭代进行,分别将细胞分为 61 (L1)、411 (L2)、1,346 (L3) 和 2,573 (L4) 簇。在每个水平上,选择高度可变的 100 kb 分箱,并使用主成分分析 (PCA) 进行降维。将 mCH 和 mCG 的重要主成分 (PC) 组合起来进行共识聚类。
We first carried out doublet removal with the help of unbiased data. The 56,843 cells after QC step 2 are mapped to the 310,605 unbiased snmC-seq cells (including predicted doublet cells). We used the highly variable features selected in the unbiased data and the PCA model fit with the unbiased data to transform the epi-retro-seq to the same dimension reduction space as the unbiased data. Then we classified the epi-retro-seq cells into either 1 of the 61 L1 clusters or the predicted doublet clusters defined in the unbiased data. The classification was carried out with the k-nearest neighbour approach described above on the PCs combining mCH and mCG. The epi-retro-seq cells assigned to each non-doublet L1 cluster were analysed in the next iteration, using the highly variable features selected in the unbiased data for the cluster and the PCA model fit with the unbiased data for the cluster. All of the predicted doublet cells in the unbiased data were added in each L1 cluster in the L2 clustering to further exclude the potential doublets. After these two iterations, the cells predicted to be doublets were removed, with 48,032 epi-retro-seq cells remaining. These cells were mapped to the 301,626 unbiased snmC-seq cells (without predicted doublets) with the same feature selection and PCA methods through the four levels, so each epi-retro-seq cell is assigned to 1 cluster at each level. The 61 L1 clusters were annotated on the basis of their dissection source and marker genes. The cell clusters representing non-neuronal cells were removed from further analyses. The cells corresponding to the IT, ET, corticothalamic and cortical inhibitory clusters in the L1 cluster annotation were used for QC step 4 as described above.
我们首先在无偏数据的帮助下进行了双峰去除。将 QC 步骤 2 后的 56,843 个细胞映射到 310,605 个无偏倚的 snmC-seq 细胞(包括预测的双细胞)。我们使用了在无偏数据中选择的高度可变的特征,并且 PCA 模型与无偏数据拟合,将 epi-retro-seq 转换为与无偏数据相同的降维空间。然后我们将 epi-retro-seq 细胞分为 61 个 L1 簇中的 1 个或无偏倚数据中定义的预测双峰簇。分类是在结合 mCH 和 mCG 的 PC 上使用上述 k 最近邻方法进行的。在下一次迭代中,使用在集群的无偏数据中选择的高度可变的特征和 PCA 模型与集群的无偏数据拟合,分析分配给每个非双峰 L1 集群的 epi-retro-seq 细胞。无偏数据中所有预测的双峰单元都添加到 L2 聚类的每个 L1 集群中,以进一步排除潜在的双峰。在这两次迭代之后,预测为双峰的细胞被去除,剩余 48,032 个 epi-retro-seq 细胞。这些细胞通过四个水平使用相同的特征选择和 PCA 方法映射到 301,626 个无偏倚的 snmC-seq 细胞(没有预测的双峰),因此每个 epi-retro-seq 细胞在每个水平被分配到 1 个簇。根据解剖来源和标记基因对 61 个 L1 簇进行注释。代表非神经元细胞的细胞簇从进一步分析中去除。如上所述,对应于 L1 簇注释中 IT、ET、皮质丘脑和皮质抑制簇的细胞用于 QC 第 4 步。
Quantification of projection neuron difference with AUROC
使用 AUROC 量化投射神经元差异
To test the similarity of two groups of cells based on DNA methylation, we trained logistic regression models to predict the group label of each cell. We compared the results using four different types of feature to predict the projection target of neurons from the same source. These include the posterior mCH level of 100-kb-bin and gene-body, and the dimension reduction results of the two matrices. A total of 50 PCs were used as dimension reduction, with unbiased snmC-seq to fit the PCA models and transform the epi-retro-seq data. We also used two methods to split the cells into training and testing sets. One used a random selection of half of the cells projecting to each target for training and the other half for testing (computational replicates); the other was based on the sex of the mice from which the cells were collected (biological replicates). After the QC steps, we have 168 source–target combinations with data from both sexes and the other 57 with cells from only one sex. Therefore, all of the comparisons of 926 target pairs could be quantified with the computational replicates, but only 516 of them could be quantified with biological replicates. We noticed significant congruence of model performance between the different features and different train/test splits (Extended Data Fig. 3a–c). The performance when using 100-kb bins was very similar to that when using gene bodies (Extended Data Fig. 3a). The performance when using raw features was slightly better than that when using PCs (Extended Data Fig. 3b). The performance when using computational replicates was significantly better than that when using biological replicates (Extended Data Fig. 3c), which was expected given that the computational replicates dismissed the heterogeneity between biological replicates and made the predictions easier. Nevertheless, the computational replicates still provided strongly correlated results to biological replicates (Extended Data Fig. 3c), which allowed the comparison between different target pairs to evaluate their epigenomic differences.
为了根据 DNA 甲基化测试两组细胞的相似性,我们训练了 logistic 回归模型来预测每个细胞的组标签。我们使用四种不同类型的特征比较了结果,以预测来自同一来源的神经元的投影目标。这些包括 100 kb-bin 和基因体的后验 mCH 水平,以及两种基质的降维结果。总共使用 50 台 PC 进行降维,使用无偏 snmC-seq 拟合 PCA 模型并转换 epi-retro-seq 数据。我们还使用了两种方法将单元格分为训练集和测试集。一个使用随机选择投射到每个目标的一半细胞进行训练,另一半用于测试(计算重复);另一个是基于收集细胞的小鼠的性别(生物重复)。在 QC 步骤之后,我们有 168 个源-靶标组合,其中包含来自两性的数据,另外 57 个包含仅来自一种性别的细胞。因此,926 个靶标对的所有比较都可以用计算重复来量化,但其中只有 516 个可以用生物学重复来量化。我们注意到不同特征和不同训练/测试拆分之间的模型性能具有显著的一致性(扩展数据图 .3a-c)。使用 100 kb bins 时的性能与使用基因体时的性能非常相似(扩展数据图 .3a). 使用原始特征时的性能略好于使用 PC 时(扩展数据图 .3b)。使用计算重复时的性能明显优于使用生物重复时的性能(扩展数据图 1)。 3c),这是意料之中的,因为计算重复消除了生物重复之间的异质性,使预测更容易。尽管如此,计算重复仍然提供了与生物学重复密切相关的结果(扩展数据图 .3c),这允许不同靶标对之间的比较以评估它们的表观基因组差异。
All of the other results in the figures were computed using the computational replicates with gene-body mCH as features. The features were filtered on the basis of average read coverage across cells before the model training. We removed the 100-kb bins and genes with <500 average CH basecalls, resulting in 23,730 bins or 9,906 genes in the model. Sci-kit learn was used for model implementation. The area under the receiver operating characteristic (AUROC) from cross-validation was used to measure the performance of the model. The higher AUROC represented the better ability of the model to present the group label, which indicated that the two groups had larger mCH differences and were more distinguishable. For computational replicates, we carried out random sampling 50 times with different seeds, and used the average AUROC as the final result.
图中的所有其他结果都是使用以基因-体 mCH 为特征的计算重复计算的。在模型训练之前,根据单元格的平均读取覆盖率对特征进行筛选。我们删除了 100 kb 的 bin 和具有 <500 平均 CH 碱基调用的基因,从而在模型中得到 23,730 个 bin 或 9,906 个基因。Sci-kit learn 用于模型实现。来自交叉验证的受试者工作特征下面积 (AUROC) 用于测量模型的性能。较高的 AUROC 代表模型呈现组标签的能力更好,这表明两组具有更大的 mCH 差异并且更易区分。对于计算重复,我们使用不同的种子进行了 50 次随机采样,并使用平均 AUROC 作为最终结果。
To test the predictability of projection targets with genes from different categories, we collected the genes from the following resources—neuropeptides and receptors: Table 1 in ref. 42 and Supplementary Fig. 16 in ref. 43; neurotransmitter receptors: Supplementary Fig. 15 in ref. 43; ion channels: Supplementary Fig. 14 in ref. 43 and the Guide to PHARMACOLOGY database (https://www.guidetopharmacology.org/DATA/targets_and_families.csv); neural projection development: Gene Ontology terms GO0031175 Neuron Projection Development and GO0050808 Synapse Organization; TFs: annotation from SCENIC+ (ref. 44). Only genes included in 9,906 genes with high CH coverage were analysed, and adding more lower-coverage genes to increase the size of gene sets did not improve the prediction performance.
为了测试不同类别基因的投射靶标的可预测性,我们从以下资源中收集了基因——神经肽和受体:参考文献 42 中的表 1 和参考文献 43 中的补充图 16;神经递质受体:参考文献 15 中的补充图 43;离子通道:参考文献 43 中的补充图 14 和药理学指南数据库 (https://www.guidetopharmacology.org/DATA/targets_and_families.csv);神经投射发育:基因本体论术语 GO0031175 神经元投射发育和 GO0050808 突触组织;TFs:来自 SCENIC+ 的注释(参考文献 44)。仅分析了 9,906 个具有高 CH 覆盖率的基因中包含的基因,添加更多低覆盖度基因以增加基因集的大小并不能提高预测性能。
Several reasons could contribute to a low prediction performance. Biological reasons would include the following. First, some neurons make projections to several targets simultaneously. These could result in the neurons being captured by several retrograde labelling experiments of different targets. It would be impossible to predict a single label with our pairwise models for this type of neuron. Second, some neurons project to different target regions but have tiny epigenetic differences. To systematically distinguish the first and second reasons, other anatomic and genetic validations are still needed.
有几个原因可能导致预测性能低下。生物学原因包括以下内容。首先,一些神经元同时向多个目标进行投射。这可能导致神经元被不同靶标的几个逆行标记实验捕获。对于这种类型的神经元,使用我们的成对模型不可能预测单个标签。其次,一些神经元投射到不同的靶区域,但具有微小的表观遗传差异。为了系统地区分第一个和第二个原因,仍然需要其他解剖学和遗传学验证。
Technical reasons would include the following. First, the contamination levels of some experiments might be relatively high, which make larger noise and hinder the models from capturing real projection differences. Second, the epigenetic differences between neurons projecting to different targets vary across replicates. Third, the sample sizes of some projections are small, which makes learning more challenging. Fourth, the models are not powerful enough to capture the complex differences between projections.
技术原因包括以下内容。首先,某些实验的污染水平可能相对较高,这会产生更大的噪声并阻碍模型捕获真实的投影差异。其次,投射到不同靶标的神经元之间的表观遗传差异因重复而异。第三,一些预测的样本量很小,这使得学习更具挑战性。第四,这些模型不够强大,无法捕捉预测之间的复杂差异。
Elimination of contaminated FANS runs in QC step 4 decreased the potential influence by the first technical reason for cortical neurons as discussed in the “Quality control” section, although there are still contaminated cells included in the dataset. The improvement in labelling efficiency and specificity would help to better solve the molecular differences between projection types. In this study, male and female mice were treated as biological replicates after removing sex chromosomes. Although methylation patterns of autosomes are similar, differences between sexes or animals might still exist. The small differences in performance between data splitting methods (based on computation or biological replicates) might suggest a less notable effect contributed by the second technical reason in those samples. To evaluate the potential limitation of the fourth technical reason, more carefully curated models, and accordingly, more samples, would be required. Thus, given all of these factors, we are generally more confident in the distinguishable target pairs when training and testing sets were split on the basis of both computational and biological replicates. The interpretation of comparisons without biological replicates and the indistinguishable pairs would need to be more careful and are not involved in the major conclusions in this manuscript. Our study aims to provide a general view across many sources and targets. A more detailed understanding of specific projections would require larger-scale profiles on those specific projection types.
正如“质量控制”部分所讨论的,在 QC 步骤 4 中消除受污染的 FANS 运行减少了皮层神经元第一个技术原因的潜在影响,尽管数据集中仍包含受污染的细胞。标记效率和特异性的提高将有助于更好地解决投射类型之间的分子差异。在这项研究中,雄性和雌性小鼠在去除性染色体后被视为生物学复制。尽管常染色体的甲基化模式相似,但性别或动物之间的差异可能仍然存在。数据拆分方法(基于计算或生物学重复)之间的微小性能差异可能表明这些样本中的第二个技术原因贡献的影响不太明显。为了评估第四个技术原因的潜在局限性,需要更仔细策划的模型,因此需要更多的样本。因此,考虑到所有这些因素,当训练集和测试集根据计算和生物重复进行拆分时,我们通常对可区分的目标对更有信心。对没有生物学重复和无法区分的对的比较的解释需要更加小心,并且不涉及本手稿中的主要结论。我们的研究旨在提供许多来源和目标的一般视图。要更详细地了解特定投影,则需要对这些特定投影类型进行更大规模的剖析。
Integration between snmC-seq, epi-retro-seq and scRNA-seq
snmC-seq、epi-retro-seq 和 scRNA-seq 之间的整合
snmC-seq and scRNA-seq data used in this study are comprehensive atlases of the whole mouse brain, so most of the cell types are expected to be present in both datasets. Therefore, the two datasets were integrated on the basis of a canonical correlation analysis (CCA) framework, which captures the shared variation between the two datasets41. Epi-retro-seq is a projection-enriched dataset that contains a subset of the cell types in the atlas, but the shared methylation modality with snmC-seq allowed it to be integrated with the comprehensive atlas with a reciprocal PCA framework. Both the epi-retro-seq and the scRNA-seq datasets were mapped to the dimension reduction space of the snmC-seq data to create a multi-modality atlas of each brain region group.
本研究中使用的 snmC-seq 和 scRNA-seq 数据是整个小鼠大脑的综合图谱,因此预计大多数细胞类型将存在于两个数据集中。因此,这两个数据集是在典型相关分析 (CCA) 框架的基础上整合的,该框架捕获了两个数据集之间的共同变化41。Epi-retro-seq 是一个投影丰富的数据集,其中包含图谱中细胞类型的子集,但与 snmC-seq 共享的甲基化模式允许它与具有互惠 PCA 框架的综合图谱集成。epi-retro-seq 和 scRNA-seq 数据集都映射到 snmC-seq 数据的降维空间,以创建每个脑区组的多模态图谱。
For each region group, we selected cells from the three datasets belonging to the dissection regions. The methylation cells in the L1 clusters corresponding to cerebellar neurons were excluded from the analysis of cerebral and brainstem regions. The RNA cells from the major classes of non-neuronal cells and immature neurons, and the subclasses of cerebellar neurons, were excluded from the analyses. The RNA cells from subclasses of medial mammillary nucleus (MM) and dorsal cochlear nucleus (DCO) were also excluded owing to the dissection differences between the two studies.
对于每个区域组,我们从属于解剖区域的三个数据集中选择了细胞。对应于小脑神经元的 L1 簇中的甲基化细胞被排除在脑和脑干区域的分析之外。来自非神经元细胞和未成熟神经元主要类别的 RNA 细胞以及小脑神经元亚类的 RNA 细胞被排除在分析之外。由于两项研究之间的解剖差异,来自内侧核 (MM) 和耳蜗背核 (DCO) 亚类的 RNA 细胞也被排除在外。
The gene expression levels of scRNA-seq cells were normalized by dividing the total unique molecular identifier (UMI) count of the cell and multiplying the average total UMI count of all cells, and then log transformed. The posterior gene-body mCH levels of snmC-seq and epi-retro-seq cells were used. The cluster-enriched genes (CEGs) were identified in each L4 cluster. We checked the variance of the mCH CEGs among the snmC-seq cells and scRNA-seq cells and used only the CEGs with mCH variance greater than 0.05 and expression variation greater than 0.005 for the analyses. The opposite of mCH levels was used for snmC-seq and epi-retro-seq data owing to the negative correlation between gene-body DNA methylation and gene expression. We fitted a PCA model with the snmC-seq cells and transformed the epi-retro-seq cells and scRNA-seq cells with the model. The PCs were normalized by the singular value of each dimension to avoid the embedding being driven by the first few PCs.
通过将细胞的总唯一分子标识符 (UMI) 计数除以所有细胞的平均总 UMI 计数,然后进行对数转换,对 scRNA-seq 细胞的基因表达水平进行归一化。使用 snmC-seq 和 epi-retro-seq 细胞的后基因-体 mCH 水平。在每个 L4 簇中鉴定出簇富集基因 (CEG)。我们检查了 snmC-seq 细胞和 scRNA-seq 细胞之间 mCH CEGs 的方差,并仅使用 mCH 方差大于 0.05 且表达变异大于 0.005 的 CEGs 进行分析。由于基因-体 DNA 甲基化与基因表达呈负相关,因此 snmC-seq 和 epi-retro-seq 数据使用与 mCH 水平相反的水平。我们用 snmC-seq 细胞拟合了 PCA 模型,并用该模型转化了 epi-retro-seq 细胞和 scRNA-seq 细胞。PC 由每个维度的奇异值进行归一化,以避免嵌入由前几个 PC 驱动。
We adopted a similar framework to that of Seurat v3 (ref. 41) for data integration by first identifying the mutual nearest neighbours as anchors between datasets, and then aligning the datasets through the anchors.
我们采用了与 Seurat v3(参考文献 41)类似的框架进行数据集成,首先将彼此最近的邻居确定为数据集之间的锚点,然后通过锚点对齐数据集。
To find anchors between snmC-seq and scRNA-seq, we first z-score scaled the mCH matrix and expression matrix of CEGs across cells, and the resulting matrices are represented as X (mC cell-by-CEG) and Y (RNA cell-by-CEG), respectively. CCA was used to find the shared low-dimensional embedding of the two datasets, solved by singular value decomposition of their dot product . U and V were normalized by dividing the L2 norm of each row, and were used to find five mutual nearest neighbours as anchors and score anchors using the same method as Seurat v3.
为了找到 snmC-seq 和 scRNA-seq 之间的锚点,我们首先对跨细胞的 CEG 的 mCH 矩阵和表达矩阵进行 z 分数缩放,所得矩阵分别表示为 X (mC cell-by-CEG) 和 Y (RNA cell-by-CEG)。CCA 用于查找两个数据集的共享低维嵌入,通过它们的点积 的奇异值分解来解决。U 和 V 通过除以每行的 L2 范数进行归一化,并用于查找五个彼此最近的邻居作为锚点,并使用与 Seurat v3 相同的方法对锚点进行评分。
The original CCA framework of Seurat (v3) is difficult to scale up to millions of cells owing to the memory bottleneck, since the mC cell-by-RNA matrix was used as the input to CCA. To handle this limitation, we randomly selected 50,000 cells from each dataset ( and ) as a reference to fit the CCA and transformed the other cells ( and ) onto the same canonical correlation space. Specifically, the canonical correlation vectors (CCVs) of and (denoted as and ) were computed by , for which and . Then the CCV of and (denoted as and ) were computed by and . The embeddings from the reference and query cells were concatenated for anchor identification.
由于内存瓶颈,Seurat (v3) 的原始 CCA 框架难以扩展到数百万个细胞,因为 mC cell-by-RNA 矩阵被用作 CCA 的输入。为了解决这一限制,我们从每个数据集 ( 和 ) 中随机选择了 50,000 个单元格作为参考来拟合 CCA,并将其他单元格 ( 和 ) 转换到相同的规范相关空间。具体来说,和 ( 表示为 和 ) 的 规范相关向量 (CCV) 由 计算,其中 和 。然后 和 的 CCV(表示为 和 )由 和 计算。 来自引用和查询单元格的嵌入被连接起来以进行锚点识别。
To find anchors between snmC-seq and epi-retro-seq, we used the snmC-seq data to fit a PCA model and use the model to transform epi-retro-seq cells to the same space and find the five nearest snmC-seq cells for each epi-retro-seq cell. Reciprocally, we fit another PCA model with the epi-retro-seq cells and transform the snmC-seq cells and find five nearest epi-retro-seq cells for each snmC-seq cell. The mutual nearest neighbours between the two datasets were used as anchors and scored using the same method as Seurat v3.
为了找到 snmC-seq 和 epi-retro-seq 之间的锚点,我们使用 snmC-seq 数据拟合 PCA 模型,并使用该模型将 epi-retro-seq 细胞转换为相同的空间,并为每个 epi-retro-seq 细胞找到五个最近的 snmC-seq 细胞。反过来,我们将另一个 PCA 模型与 epi-retro-seq 细胞拟合并转化 snmC-seq 细胞,并为每个 snmC-seq 细胞找到五个最近的 epi-retro-seq 细胞。两个数据集之间的互近邻被用作锚点,并使用与 Seurat v3 相同的方法进行评分。
The PCs derived from the previous step were then integrated together using the same method as Seurat v3 through these anchors. This integration step projects the PCs of epi-retro-seq and scRNA-seq (query) to the PCs of the snmC-seq (reference) while keeping the PCs of the reference dataset unchanged. The resulting PCs from the three datasets were used for t-SNE visualization and k-nearest neighbour (k = 25) graph construction with Euclidean distance. The joint clustering was carried out with the Leiden algorithm on the graph using a resolution of 1.0.
然后,使用与 Seurat v3 相同的方法通过这些锚点将上一步衍生的 PC 集成在一起。此集成步骤将 epi-retro-seq 和 scRNA-seq (查询)的 PC 投影到 snmC-seq (参考)的 PC,同时保持参考数据集的 PC 不变。来自三个数据集的 PC 用于 t-SNE 可视化和具有欧几里得距离的 k 最近邻 (k = 25) 图构造。联合聚类是使用 Leiden 算法在图形上以 1.0 的分辨率进行的。
The quality of the integration analysis was evaluated from two aspects. First, we visualized the different modalities in the co-embedding space (Extended Data Fig. 4 left). The local neighbourhoods of the co-embedding usually contain cells from all modalities, suggesting a good mixture between the three datasets after integration. Second, we computed the proportion of cells in each mC cluster (Extended Data Fig. 4 middle) or RNA cluster (Extended Data Fig. 4 right) assigned to each cluster defined on the co-embedding space (co-cluster). As we used the highest granularity of clustering from individual modalities (original cluster), the co-clusters were usually larger than the original clusters. We therefore used the proportion of original clusters rather than the proportion of co-clusters, to demonstrate that almost all original clusters are included in one co-cluster with low ambiguity. The strongest signals align on the diagonals suggesting that the co-embedding preserved the cluster structures that were originally present within each modality. Further evidence of integration quality was suggested by the downstream analyses, for which highly consistent cell-type specificity of marker-gene expression and gene-body mCH were observed (Figs. 3f,g and 4e,f and Extended Data Fig. 9a).
从两个方面评价整合分析的质量。首先,我们可视化了共嵌入空间中的不同模态(扩展数据图 .4 左)。共嵌入的局部邻域通常包含来自各种模态的细胞,这表明整合后三个数据集之间具有良好的混合。其次,我们计算了每个 mC 簇中细胞的比例(扩展数据图 D)。4 中间)或 RNA 簇(扩展数据图4 右)分配给在共嵌入空间 (co-cluster) 上定义的每个集群。由于我们使用了来自单个模态(原始集群)的最高聚类粒度,因此协聚类通常比原始聚类大。因此,我们使用原始聚类的比例而不是协聚类的比例来证明几乎所有原始聚类都包含在一个低歧义的协聚类中。最强的信号在对角线上对齐,表明共嵌入保留了最初存在于每种模态中的簇结构。下游分析进一步证明了整合质量,其中观察到标记基因表达和基因-体 mCH 的高度一致的细胞类型特异性(图3f,g 和 4e,f 和扩展数据图9a)。
Cluster associated with projection
与投影关联的群集
For neurons projecting to each target within one source, we computed the proportion of these neurons in each joint Leiden cluster. The clusters with >5% of the cells were considered as associated with the projection. The clusters associated with at least one projection are shown in the heatmaps of Figs. 3–5 and Extended Data Fig. 5. The values in the heatmaps represent the proportion of projection neurons in each cluster, z-scored within each cluster across the projection targets. We used the Fisher exact test to quantify whether one projection has a similar proportion of cells in each cluster compared with all other projections. One-sided tests were used to select odds ratios greater than 1 (projection cells enriched in one cluster compared with other projections). FDR values < 0.01 are labelled in the heatmaps in Figs. 3–5 and Extended Data Fig. 5. We also used the Fisher exact test to quantify whether the male and female samples have a similar proportion of cells in each cluster, when the samples of both sexes passed the QC described above. Two-sided tests were carried out and FDR values < 0.01 are labelled in the heatmaps in Extended Data Fig. 5.
对于投射到一个来源内每个目标的神经元,我们计算了这些神经元在每个关节 Leiden 集群中的比例。具有 >5% 个像元的聚类被认为与投影相关。与至少一个投影相关的集群显示在 Figs 的热图中。图 3–5 和扩展数据图5. 热图中的值表示每个集群中投影神经元的比例,每个集群内跨投影目标的 z 分数。我们使用 Fisher 精确检验来量化与所有其他投影相比,一个投影在每个聚类中的像元比例是否相似。使用单侧检验选择大于 1 的比值比(与其他预测相比,在一个簇中富集的投影细胞)。< 0.01 的 FDR 值在图 3-5 和扩展数据图 3 的热图中标记。5. 我们还使用 Fisher 精确检验来量化当两性样本通过上述 QC 时,雄性和雌性样本在每个簇中是否具有相似比例的细胞。进行了双面测试,FDR 值< 0.01 标记在扩展数据图 5 的热图中。
In general, there are two intuitive ways to quantify the enrichment of projection neurons in a cluster. One is to directly find the clusters with a high absolute proportion of epi-retro-seq neurons projecting to a target. The other is to find clusters captured at a significantly higher frequency in the projection-enriched data relative to the unbiased data. The two methods each have their advantages and shortcomings. For example, the contaminated cells from inaccurate labelling or gating are likely to have a similar distribution across clusters to unbiased profiling. So a comparison using unbiased data as a control might help exclude the contaminated clusters better. However, if most of the neurons from a projection type are in the clusters that are originally abundant cell types in the source, by comparing with unbiased data, we would miss the predominant clusters making the projection.
一般来说,有两种直观的方法可以量化集群中投射神经元的富集。一种是直接找到投射到目标的 epi-retro-seq 神经元绝对比例高的簇。另一种方法是在投影丰富的数据中查找相对于无偏数据以明显更高的频率捕获的聚类。这两种方法各有优缺点。例如,由于标记或门控不准确而受污染的细胞可能在簇中具有与无偏倚分析相似的分布。因此,使用无偏倚数据作为对照进行比较可能有助于更好地排除受污染的集群。但是,如果来自投影类型的大多数神经元位于源中最初丰富的细胞类型的簇中,则通过与无偏数据进行比较,我们将错过进行投影的主要簇。
In this manuscript, we used the absolute proportions but not the relative ones to the unbiased data owing to the different profiling strategies between the two datasets. Although the epi-retro-seq samples and unbiased snmC-seq samples were dissected in the same way, we pooled the different dissections into the 32 different sources to carry out FANS and sequencing for epi-retro-seq, so that the proportion of cells from different dissection regions of the same source is likely to follow their proportions in the mouse brain. However, the unbiased snmC-seq profiled all of the dissection regions separately and sequenced the same number of cells in each dissection, which manually amplified the proportion of cells from the smaller or sparser dissection regions relative to the larger or denser ones, and limited the power to estimate the real proportion of neurons in each cluster from the sources.
在这份手稿中,由于两个数据集之间的分析策略不同,我们使用了无偏数据的绝对比例,但没有使用相对比例。尽管 epi-retro-seq 样本和无偏倚的 snmC-seq 样本以相同的方式解剖,但我们将不同的解剖整合到 32 个不同的来源中,以进行 epi-retro-seq 的 FANS 和测序,因此来自同一来源不同解剖区域的细胞比例很可能遵循它们在小鼠大脑中的比例。然而,无偏倚的 snmC-seq 分别分析了所有解剖区域,并对每个解剖中相同数量的细胞进行了测序,这手动放大了来自较小或较稀疏解剖区域的细胞比例相对于较大或较密集的解剖区域,并限制了从来源估计每个簇中神经元的实际比例的能力。
As we pooled the two mice of the same sex before sequencing, we do not have biological replicates to study the different distribution across clusters of projection neurons from different sexes. For the identification of projection-enriched clusters and their differences between sexes, we used Fisher exact tests to find the proportion difference that treats each single cell as an independent sample but does not consider the consistency between biological replicates. This could lead to false discovery, so we consider the projection-enriched clusters with no significant sexual proportion differences as more confident ones. We also tested general linear models with binomial dependent variables and use biological replicates as a random effect. However, probably owing to the number of replicates being too small (1 or 2), the limited power for accessing random effects resulted in low detection power and the test was highly biased to identifying small clusters. Therefore, our dataset provides a general resource across the whole brain suggesting projection-associated cell types, and more biological replicates are needed to validate the patterns and investigate the sexual differences.
由于我们在测序前合并了两只同性别的小鼠,因此我们没有生物学重复来研究来自不同性别的投射神经元簇之间的不同分布。为了鉴定富含投射的簇及其性别之间的差异,我们使用 Fisher 精确检验来寻找将每个单个细胞视为独立样本但不考虑生物学重复之间的一致性的比例差异。这可能会导致错误的发现,因此我们认为没有显著性别比例差异的投影丰富集群是更可信的集群。我们还测试了具有二项式因变量的一般线性模型,并使用生物重复作为随机效应。然而,可能是由于重复数太少(1 或 2 个),访问随机效应的能力有限,导致检测能力低,并且测试高度偏倚于识别小集群。因此,我们的数据集提供了整个大脑的通用资源,表明与投射相关的细胞类型,并且需要更多的生物学重复来验证模式并研究性别差异。
It is also worth noting that our integration and clustering strategies did not consider the projection target labels of neurons. Therefore, the granularity and boundary of unbiased clusters could be different from the actual projection-associated cell types. For example, some clusters may have only a small fraction enriched for certain projection neurons, whereas some clusters may have very similar projection enrichment patterns but still split into several in our analyses. Improved methods for clustering including the information of both molecular feature embedding and projection labels could further expand our understanding of the association between molecular cell types and neural projections.
还值得注意的是,我们的整合和聚类策略没有考虑神经元的投影目标标签。因此,无偏聚类的粒度和边界可能与实际的投影相关像元类型不同。例如,一些集群可能只有一小部分针对某些投影神经元进行了富集,而一些集群可能具有非常相似的投影富集模式,但在我们的分析中仍分为几个。改进的聚类方法,包括分子特征嵌入和投影标记的信息,可以进一步扩大我们对分子细胞类型和神经投射之间关联的理解。
Classification of MERFISH cells into major brain regions and cell clusters
将 MERFISH 细胞分为主要大脑区域和细胞簇
The MERFISH experiments were conducted as described in ref. 17, including the gene panel design, tissue preparation, imaging, data processing and annotation. The dataset includes two sagittal slices (S1 and S2, with S1 being more lateral and S2 being more medial) and 14 coronal slices (C2, C4, C6, C8, C10, C12 and C14, roughly corresponding to slices 2, 4, 6, 8, 10, 12 and 14 in Extended Data Fig. 1, with two replicates for each slice, represented as R1 and R2). The same naming of slices was used throughout this manuscript (Figs. 3e, 4d and 5c and Extended Data Fig. 6).
MERFISH 实验按照参考文献 17 中的描述进行,包括基因面板设计、组织制备、成像、数据处理和注释。该数据集包括两个矢状切片(S1 和 S2,其中 S1 更外侧,S2 更内侧)和 14 个冠状切片(C2、C4、C6、C8、C10、C12 和 C14,大致对应于扩展数据图 2 中的切片 2、4、6、8、10、12 和 14。1,每个切片有两个重复,表示为 R1 和 R2)。本手稿中使用相同的切片命名(图 .3e、4d 和 5c 以及扩展数据图6).
The MERFISH cells were classified into subclasses and brain region groups by integration with scRNA-seq data. The 489 autosomal genes that overlapped between scRNA-seq and MERFISH datasets were used. We fitted a PCA model with the scRNA-seq cells and transformed the MERFISH cells with the model. The PCs were normalized by the singular value of each dimension. The cell-by-gene matrices were z-score normalized across cells within each dataset, and CCA was used to find anchors between the two datasets. We used 50,000 cells to fit the CCA and transformed the other cells as described above. The transformed PCs of MERFISH cells were then aligned to the PCs of scRNA-seq cells to derive a co-embedding between the two datasets. This co-embedding was used for label transfer of cell subclasses from scRNA-seq data to MERFISH data, considering 25 neighbouring scRNA-seq cells for each MERFISH cell.
通过与 scRNA-seq 数据整合,将 MERFISH 细胞分为亚类和脑区组。使用了 scRNA-seq 和 MERFISH 数据集之间重叠的 489 个常染色体基因。我们用 scRNA-seq 细胞拟合了 PCA 模型,并用该模型转化了 MERFISH 细胞。PC 由每个维度的奇异值进行归一化。每个数据集中的细胞对 z 分数进行归一化,并使用 CCA 查找两个数据集之间的锚点。我们使用 50,000 个细胞来拟合 CCA,并如上所述转化其他细胞。然后将 MERFISH 细胞的转化 PC 与 scRNA-seq 细胞的 PC 进行比对,以得出两个数据集之间的共嵌入。考虑到每个 MERFISH 细胞有 25 个相邻的 scRNA-seq 细胞,这种共嵌入用于将细胞亚类从 scRNA-seq 数据标记转移到 MERFISH 数据。
The cells classified as non-neuronal and immature neuronal subclasses were excluded owing to lack of regional specificity, and the rest of cells from the two datasets were integrated again with the procedures described above to transfer the label of 14 brain region groups from the scRNA-seq neurons to the MERFISH neurons. The initial label assignment is noisy. Therefore, a smoothing step was carried out to refine the region group assignment. Specifically, for each MERFISH cell i, we found its 25 neighbours on the same slice (denoted as ) based on the spatial coordinates, and used to represent the corresponding distances between i and its jth neighbour . Similarly, we found the 25 scRNA-seq neighbours for each MERFISH cell based on the integration, and used to represent the distances. The distance matrices were transformed as described in the label transfer section, and the final spatial labels were transferred from the 25 RNA neighbours of each of the 25 spatial neighbours (625 scRNA-seq cells in total) to 1 MERFISH cell. The weight between the MERFISH cell i and the kth scRNA-seq neighbour of its jth spatial neighbour was computed as . is row normalized and used as weights for label transfer as described in previous sections.
由于缺乏区域特异性,分类为非神经元和未成熟神经元亚类的细胞被排除在外,来自两个数据集的其余细胞再次与上述程序整合,以将 14 个脑区组的标记从 scRNA-seq 神经元转移到 MERFISH 神经元。初始标签分配是干扰的。因此,执行了平滑步骤以优化区域组分配。具体来说,对于每个 MERFISH 单元格 i,我们根据空间坐标在同一个切片(表示为 )上找到它的 25 个邻居,并用来 表示 i 和它的第 j个邻居之间的相应距离。 同样,我们根据积分找到了每个 MERFISH 细胞 的 25 个 scRNA-seq 邻居,并用于 表示距离。按照标签转移部分的描述转换距离矩阵,并将最终空间标签从 25 个空间邻居中每个空间邻居的 25 个 RNA 邻居(总共 625 个 scRNA-seq 细胞)转移到 1 个 MERFISH 细胞。MERFISH 细胞 i 与其第 j个空间邻居的第 k个 scRNA-seq 邻居之间的权重计算为 。 行规范化并用作标签传输的权重,如前面部分所述。
We note that this could also be achieved through registration of MERFISH DAPI images to the common coordinate framework. However, our companion works demonstrated that the procedure is relatively challenging and it is important to use cell types with known locations as landmarks to refine the registration.
我们注意到,这也可以通过将 MERFISH DAPI 图像注册到公共坐标框架来实现。然而,我们的配套工作表明,该程序相对具有挑战性,使用具有已知位置的细胞类型作为标志来改进注册非常重要。
Finally, the neurons from each region group were selected and integrated with the scRNA-seq cells from the same region group, using the same procedure as described above. The mC–RNA co-cluster labels were transferred from the scRNA-seq cell to the MERFISH cells. The MERFISH cells assigned to the MM and DCO subclasses in the last step were also excluded as those clusters were not included in the co-cluster analysis as described in the previous section.
最后,使用与上述相同的程序,选择来自每个区域组的神经元并将其与来自同一区域组的 scRNA-seq 细胞整合。mC-RNA 共簇标记从 scRNA-seq 细胞转移到 MERFISH 细胞。在上一步中分配给 MM 和 DCO 亚类的 MERFISH 细胞也被排除在外,因为这些簇不包括在上一节所述的共簇分析中。
Comparison with Act-seq 与 Act-seq 的比较
The 10x scRNA-seq data in ref. 7 were downloaded from Mendeley Data (https://data.mendeley.com/datasets/ypx3sw2f7c/3), and are referred to as Act-seq in this section and Extended Data Fig. 7. The dataset contains 168,877 cells in total, among which 78,476 were labelled as neurons and were used to integrate to the unbiased scRNA-seq data for hypothalamic neurons, using the 5,314 CEGs of 1,891 L3 scRNA-seq clusters. PCA was fitted with scRNA-seq data and the Act-seq data were transformed. The CCA framework was used to find anchors between the two datasets, and the transformed PCs of Act-seq were aligned to the scRNA-seq PCs. We used the label transfer method described in the previous section to transfer the mC–RNA co-cluster labels from scRNA-seq cells to the Act-seq cells, considering five neighbouring scRNA-seq cells for each Act-seq cell. The Act-seq cells with Fos expression level > 0 were considered to be Fos+ cells, and the proportions of Fos+ cells were compared between control and each behaviour using one-sided Fisher exact tests. The Fos expression levels were compared with one-sided Wilcoxon rank-sum tests.
参考文献 7 中的 10x scRNA-seq 数据是从 Mendeley Data (https://data.mendeley.com/datasets/ypx3sw2f7c/3) 下载的,在本节和 Extended Data Fig.7. 该数据集总共包含 168,877 个细胞,其中 78,476 个被标记为神经元,并使用 1,891 个 L3 scRNA-seq 簇的 5,314 个 CEG 整合到下丘脑神经元的无偏 scRNA-seq 数据中。用 scRNA-seq 数据拟合 PCA,并转换 Act-seq 数据。使用 CCA 框架寻找两个数据集之间的锚点,并将 Act-seq 转换后的 PC 与 scRNA-seq PC 对齐。我们使用上一节中描述的标签转移方法将 mC-RNA 共簇标记从 scRNA-seq 细胞转移到 Act-seq 细胞,考虑每个 Act-seq 细胞的五个相邻 scRNA-seq 细胞。Fos 表达水平> 0 的 Act-seq 细胞被认为是 Fos + 细胞,并使用单侧 Fisher 精确检验比较对照和每种行为之间 Fos + 细胞的比例。将 Fos 表达水平与单侧 Wilcoxon 秩和检验进行比较。
Only the 23,345 Act-seq cells from 16 ventromedial HY (VMH) neuron clusters were considered as ventrolateral VMH cells (VMHvl) and were used for the behaviour-association studies in ref. 7. However, almost none of them correspond to projection-associated clusters in our data (Extended Data Fig. 7a–c). We further compared our projection-associated clusters with all of the neuron clusters profiled in ref. 7 and note that five clusters have corresponding clusters in the Act-seq data (Extended Data Fig. 7d, in red). Among them, clusters 4 and 64 showed weak but significant increases in proportions of Fos+ cells labelled during certain behaviours (Extended Data Fig. 7f).
只有来自 16 个腹内侧 HY (VMH) 神经元簇的 23,345 个 Act-seq 细胞被认为是腹外侧 VMH 细胞 (VMHvl),并用于参考文献 7 中的行为关联研究。然而,它们几乎没有一个对应于我们数据中的投影相关集群(扩展数据图 .7a-c)。我们进一步将我们的投影相关集群与参考文献 7 中分析的所有神经元集群进行了比较,并注意到五个集群在 Act-seq 数据中具有相应的集群(扩展数据图 .7d,红色)。其中,聚类 4 和 64 显示在某些行为期间标记的 Fos+ 细胞比例微弱但显著增加(扩展数据图 .7f)。
The generally weak associations between projection-associated and behaviour-associated clusters are probably due to the small overlap between the brain regions profiled in the two datasets, particularly the under representation of VMHvl neurons in epi-retro-seq data. Additionally, because there were far fewer cells profiled in epi-retro-seq versus Act-seq, the granularity of clusters used for projection association and behaviour association is different; this difference is particularly pronounced in VMHvl for which >10 times more cells were used for the behaviour-association study (Extended Data Fig. 7c–e). Therefore, further increasing the size of datasets to achieve higher granularity of cell typing in specific regions of interest could facilitate further association between molecular types with projections and behaviours. Our study aimed at a comprehensive view of a large number of projections across the whole brain and focused on targets that do not seem to receive strong input from VMH. This apparently limited the data overlap between these and limited the ability to make direct comparisons between studies.
投影相关集群和行为相关集群之间的关联通常较弱,这可能是由于两个数据集中描述的大脑区域之间的小重叠,特别是 VMHvl 神经元在 epi-retro-seq 数据中的代表性不足。此外,由于 epi-retro-seq 与 Act-seq 中分析的细胞要少得多,因此用于投影关联和行为关联的聚类的粒度不同;这种差异在 VMHvl 中尤为明显,其中 >10 倍的细胞用于行为关联研究(扩展数据图 .7c-e)。因此,进一步增加数据集的大小以在特定感兴趣区域实现更高粒度的细胞分型可以促进分子类型与投射和行为之间的进一步关联。我们的研究旨在全面了解整个大脑的大量投射,并专注于似乎没有从 VMH 获得强烈输入的目标。这显然限制了这些之间的数据重叠,并限制了在研究之间进行直接比较的能力。
Comparison with retro-seq
与 retro-seq 的比较
The scRNA-seq data in ref. 8 were downloaded from the Gene Expression Omnibus with the identifier GSE133912. The retro-seq data were integrated to the unbiased scRNA-seq data for thalamic neurons, using the 5,404 CEGs of 1,128 L3 scRNA-seq clusters. PCA was fitted with scRNA-seq data and retro-seq data were transformed. The CCA framework was used to find anchors between the two datasets, and the transformed PCs of retro-seq were aligned to the scRNA-seq PCs. To compare the distribution of retro-seq cells and epi-retro-seq cells across thalamic cell clusters, we used the label transfer method described in the previous section to transfer the mC–RNA co-cluster label and the joint t-SNE coordinates from scRNA-seq cells to the retro-seq cells, considering five neighbouring scRNA-seq cells for each retro-seq cell.
参考文献 8 中的 scRNA-seq 数据是从基因表达综合中下载的,标识符为 GSE133912。使用 1,128 个 L3 scRNA-seq 簇的 5,404 个 CEG,将 retro-seq 数据整合到丘脑神经元的无偏 scRNA-seq 数据中。PCA 拟合 scRNA-seq 数据,并转换 retro-seq 数据。使用 CCA 框架在两个数据集之间寻找锚点,并将 retro-seq 的转换 PC 与 scRNA-seq PC 对齐。为了比较 retro-seq 细胞和 epi-retro-seq 细胞在丘脑细胞簇中的分布,我们使用上一节中描述的标签转移方法将 mC-RNA 共簇标记和联合 t-SNE 坐标从 scRNA-seq 细胞转移到 retro-seq 细胞,考虑到每个 retro-seq 细胞有五个相邻的 scRNA-seq 细胞。
DEGs DEG
The gene expression level of each single cell was normalized by the total UMI count of the cell and log transformed. We carried out pairwise comparisons between clusters associated with projection neurons. For each cluster pair, the P values were derived with two-sided Wilcoxon rank-sum tests, and the fold change is computed as the ratio between the average expression level across cells in the two clusters. The genes with absolute value of log2 fold change greater than 1 and FDR (Benjamini–Hochberg procedure) values smaller than 0.01 were considered as differentially expressed. The DEGs from all cluster pairs were merged to generate the heatmaps in Figs. 3 and 4. Only the top 100 DEGs ranked by FDRs were used if there were more than 100 DEGs identified between a pair of clusters.
通过细胞的总 UMI 计数和对数转换对每个单个细胞的基因表达水平进行标准化。我们在与投射神经元相关的集群之间进行了成对比较。对于每个聚类对,P 值是通过双侧 Wilcoxon 秩和检验得出的,倍数变化计算为两个聚类中细胞的平均表达水平之间的比率。对数2 倍变化绝对值大于 1 且 FDR (Benjamini-Hochberg 程序) 值小于 0.01 的基因被认为是差异表达的。将所有集群对的 DEG 合并以生成图 1 中的热图。3 和 4.如果在一对集群之间识别出超过 100 个 DEG,则仅使用 FDR 排名前 100 的 DEG。
DMRs and association with genes
DMR 和与基因的关联
The unbiased snmC-seq cells from each mC–RNA co-cluster were merged to generate pseudobulk methylation profiles. The epi-retro-seq cells were not used owing to the different genome backgrounds of the mice to avoid confounding results. DMRs were identified within each region group between clusters using ALLCools. We then calculated the PCC between DMR mCG and gene mCH fraction. We shuffled the DMRs and genes within each sample to calculate the null PCC and estimate FDR. We filtered DMR–target edges with FDR < 0.01.
合并来自每个 mC-RNA 共簇的无偏倚 snmC-seq 细胞以生成假大量甲基化谱。由于小鼠的基因组背景不同,因此未使用 epi-retro-seq 细胞,以避免混淆结果。使用 ALLCools 在集群之间的每个区域组内识别 DMR。然后我们计算了 DMR mCG 和基因 mCH 分数之间的 PCC。我们对每个样本中的 DMR 和基因进行洗牌,以计算零 PCC 并估计 FDR。我们用 FDR < 0.01 过滤了 DMR 目标边缘。
TF motif enrichment TF 基序富集
We used an ensemble motif database from SCENIC+ (ref. 44), which contained 49,504 motif position weight matrices (PWMs) collected from 29 sources. Redundant motifs (highly similar PWMs) were combined into 8,045 motif clusters through clustering based on PWM distances calculated by TOMTOM45 by the SCENIC+ authors. Each motif cluster was annotated with one or more mouse TF genes. To calculate motif occurrence on DMRs, we used the Cluster-Buster46 implementation in SCENIC+, which scanned motifs in the same cluster together with hidden Markov models.
我们使用了来自 SCENIC+ 的集成基序数据库(参考文献 44),其中包含从 29 个来源收集的 49,504 个基序位置权重矩阵 (PWM)。冗余基序(高度相似的 PWM)通过基于 SCENIC+ 作者 TOMTOM45 计算的 PWM 距离的聚类组合成 8,045 个基序簇。每个基序簇都用一个或多个小鼠 TF 基因注释。为了计算 DMR 上的模体出现,我们使用了 SCENIC+ 中的 Cluster-Buster46 实现,它扫描了同一簇中的模体以及隐藏的马尔可夫模型。
Within each region group, we assign hypo-DMRs to each cluster if the mCG level of a DMR in the cluster is below the 10th quantile of all DMRs from the region group and below the 10th quantile of the mCG level of this DMR in all clusters from the region group. To carry out motif enrichment analysis, we used the recovery-curve-based cisTarget algorithm44. In brief, the cisTarget algorithm performed motif enrichment on the hypo-DMRs of each cluster by calculating the area under the recovery curve (AUC) for each motif, which is further normalized on the basis of all other motifs in the collection to calculate a normalized enrichment score. We used the cutoff AUC > 0.01 and normalized enrichment score > 3 to select enriched motifs. The TF-encoding genes shown in Figs. 3i and 4i were additionally required to have expression level > 0 and normalized mCH level < 1 in at least one cluster that its motif enriched in, to select the TFs that are likely to be expressed among a family of TFs showing the same motif enrichment scores.
在每个区域组中,如果区域中 DMR 的 mCG 水平低于区域组中所有 DMR 的第 10 个四分位数,并且低于该 DMR 的第 10 个四分位数,则为每个集群分配低 DMR。为了进行基序富集分析,我们使用了基于恢复曲线的 cisTarget 算法44。简而言之,cisTarget 算法通过计算每个基序的恢复曲线下面积 (AUC) 对每个簇的低 DMR 进行基序富集,该基序根据集合中的所有其他基序进一步归一化以计算归一化富集分数。我们使用 > 0.01 的临界 AUC 和 > 3 的标准化富集评分来选择富集基序。图 1 所示的 TF 编码基因。3i 和 4i 还要求在其基序富集的至少一个簇中具有> 0 的表达水平和标准化 mCH < 1 水平,以选择可能在显示相同基序富集分数的 TFs 家族中表达的 TFs。
GRN analysis GRN 分析
GRNs were constructed in each major brain region, with genes (TF and non-TF) and DMRs as nodes and three types of edge. The GRNs were summarized into many triplet combinations, each of which contains a TF, a DMR, a target gene (including TFs) and edges between each other. In this analysis, we include only the TFs whose motif is significantly enriched in hypo-DMRs of any cluster in the brain region (as described in the “TF motif enrichment” section). We use only a union set of genes with absolute log2 fold change > 1 and FDR < 0.01 (described in the section “DEGs”) in any pairwise comparisons between clusters. The weights of edges were computed on the basis of the PCC between nodes across clusters, and only the edges showing significant correlation in the shuffle tests (described in the “DMRs and association with genes” section) were kept. Before computation of the PCC, all data were quantile normalized within each cluster (across features). The edges include the following three categories: TF–DMR edges, connected if the PCC between TF expression and mCG level of DMRs is significant (FDR < 0.01) and the TF has a binding site in the DMR predicted by Cluster-Buster; TF–gene edges, connected if the PCC between TF expression and target gene expression is significant (FDR < 0.01); and DMR–gene edges, connected if the PCC between mCG level of DMRs and target gene expression is significant and the distance between gene transcription start site and the DMR is within 1 megabase.
在每个主要脑区构建 GRN,以基因 (TF 和非 TF) 和 DMRs 为节点和三种类型的边缘。将 GRNs 总结为许多三联体组合,每个组合都包含一个 TF、一个 DMR、一个靶基因(包括 TFs)和彼此之间的边缘。在此分析中,我们仅包括其基序在脑区任何簇的低 DMR 中显着富集的 TF(如“TF 基序富集”部分所述)。在簇之间的任何成对比较中,我们只使用具有绝对对数2 倍变化> 1 且 FDR < 0.01 的基因联合集(在“DEGs”一节中描述)。边的权重是根据跨集群节点之间的 PCC 计算的,并且只保留在随机播放测试中显示出显着相关性的边(在 “DMR 和与基因的关联 ”一节中描述)。在计算 PCC 之前,所有数据在每个集群内(跨特征)进行分位数归一化。这些边缘包括以下三类:TF-DMR 边缘,如果 TF 表达与 DMR 的 mCG 水平之间的 PCC 显著 (FDR < 0.01) 并且 TF 在 Cluster-Buster 预测的 DMR 中具有结合位点,则连接;如果 TF 表达和靶基因表达之间的 PCC 显著 (FDR < 0.01),则连接 TF 基因边缘;和 DMR-基因边缘,如果 DMR 的 mCG 水平与靶基因表达之间的 PCC 显著且基因转录起始位点与 DMR 之间的距离在 1 兆碱基以内,则连接。
Note that because the GRNs were constructed based only on correlations between data modalities and the binding motifs, they inevitably capture indirect and false-positive relationships. Perturbation experiments would be necessary to validate the connections between cis or trans regulators and target genes.
请注意,由于 GRN 仅基于数据模态和结合基序之间的相关性构建,因此它们不可避免地会捕获间接和假阳性关系。扰动实验对于验证顺式或反式调节因子与靶基因之间的联系是必要的。
Reporting summary 报告摘要
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
有关研究设计的更多信息,请参阅本文链接的 Nature Portfolio Reporting Summary。
Data availability 数据可用性
Raw and processed data are available at the Gene Expression Omnibus under accession code GSE230782. Processed data can be explored on our web portal: http://neomorph.salk.edu/epiretro. Other datasets used in this study include scRNA-seq (https://portal.brain-map.org/atlases-and-data/bkp/abc-atlas), snmC-seq and MERFISH (https://mousebrain.salk.edu/download), Act-seq (https://doi.org/10.17632/ypx3sw2f7c.3) and retro-seq (GSE133912). The mm10 genome was downloaded from https://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips/.
原始数据和处理后的数据可在 Gene Expression Omnibus 的登录代码 GSE230782 下获得。可以在我们的门户网站上浏览处理的数据:http://neomorph.salk.edu/epiretro。本研究中使用的其他数据集包括 scRNA-seq (https://portal.brain-map.org/atlases-and-data/bkp/abc-atlas)、snmC-seq 和 MERFISH (https://mousebrain.salk.edu/download)、Act-seq (https://doi.org/10.17632/ypx3sw2f7c.3) 和 retro-seq (GSE133912)。mm10 基因组是从 https://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips/ 下载的。
Code availability 代码可用性
The code for all of the analyses can be found at https://github.com/zhoujt1994/EpiRetroSeq2023.git.
所有分析的代码都可以在 https://github.com/zhoujt1994/EpiRetroSeq2023.git 中找到。
References 引用
Armand, E. J., Li, J., Xie, F., Luo, C. & Mukamel, E. A. Single-cell sequencing of brain cell transcriptomes and epigenomes. Neuron 109, 11–26 (2021).
Armand, E. J., Li, J., Xie, F., Luo, C. & Mukamel, E. A. 脑细胞转录组和表观基因组的单细胞测序。神经元109, 11–26 (2021)。Zhang, Z. et al. Epigenomic diversity of cortical projection neurons in the mouse brain. Nature 598, 167–173 (2021).
Zhang, Z. 等人。小鼠大脑中皮质投射神经元的表观多样性。自然598, 167–173 (2021)。Chen, X. et al. High-throughput mapping of long-range neuronal projection using in situ sequencing. Cell 179, 772–786 (2019).
Chen, X. 等人。使用原位测序对长距离神经元投射进行高通量映射。单元格179, 772–786 (2019)。Sun, Y.-C. et al. Integrating barcoded neuroanatomy with spatial transcriptional profiling enables identification of gene correlates of projections. Nat. Neurosci. 24, 873–885 (2021).
孙,Y.-C.等。将条形码神经解剖学与空间转录分析相结合,可以识别投射的基因相关性。Nat. 神经科学。24, 873–885 (2021 年)。Economo, M. N. et al. Distinct descending motor cortex pathways and their roles in movement. Nature 563, 79–84 (2018).
Economo, MN 等人。不同的下降运动皮层通路及其在运动中的作用。自然563, 79–84 (2018)。Kim, E. J. et al. Extraction of distinct neuronal cell types from within a genetically continuous population. Neuron 107, 274–282 (2020).
Kim, EJ 等人。从遗传连续的群体中提取不同的神经元细胞类型。神经元107, 274–282 (2020)。Kim, D.-W. et al. Multimodal analysis of cell types in a hypothalamic node controlling social behavior. Cell 179, 713–728 (2019).
金 D.-W.等。控制社会行为的下丘脑淋巴结细胞类型的多模态分析。细胞179, 713–728 (2019)。Phillips, J. W. et al. A repeated molecular architecture across thalamic pathways. Nat. Neurosci. 22, 1925–1935 (2019).
Phillips, JW 等人。跨丘脑途径的重复分子结构。Nat. 神经科学。22, 1925–1935 (2019)。Mo, A. et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86, 1369–1384 (2015).
Mo, A. 等人。哺乳动物大脑中神经元多样性的表观基因组特征。神经元86, 1369–1384 (2015)。Luo, C. et al. Single nucleus multi-omics identifies human cortical cell regulatory genome diversity. Cell Genomics 2, 100107 (2022).
Luo, C. 等人。单核多组学鉴定人类皮质细胞调节基因组多样性。细胞基因组学2, 100107 (2022)。Lister, R. et al. Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905 (2013).
Lister, R. 等人。哺乳动物大脑发育过程中的整体表观基因组重构。科学341, 1237905 (2013)。Tervo, D. G. R. et al. A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92, 372–382 (2016).
Tervo, D. G. R. 等人。设计型 AAV 变体允许对投射神经元进行有效的逆行访问。神经元92, 372–382 (2016)。Wang, Q. et al. The Allen Mouse Brain Common Coordinate Framework: a 3D reference atlas. Cell 181, 936–953 (2020).
Wang, Q. 等人。Allen Mouse Brain Common Coordinate Framework:3D 参考图谱。细胞181, 936–953 (2020)。Luo, C. et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604 (2017).
Luo, C. 等人。单细胞甲基化组识别哺乳动物皮层中的神经元亚型和调节元件。科学357, 600–604 (2017)。Luo, C. et al. Robust single-cell DNA methylome profiling with snmC-seq2. Nat. Commun. 9, 3824 (2018).
Luo, C. 等人。使用 snmC-seq2 进行稳健的单细胞 DNA 甲基化组分析。Nat. Commun.9, 3824 (2018 年)。Tian, W. et al. Single-cell DNA methylation and 3D genome architecture in the human brain. Science 382, eadf5357 (2023).
Tian, W. 等人。人脑中的单细胞 DNA 甲基化和 3D 基因组结构。 科学382, eadf5357 (2023)。Liu, H. et al. Single-cell DNA methylome and 3D multi-omic atlas of the adult mouse brain. Nature https://doi.org/10.1038/s41586-023-06805-y (2023).
Liu, H. 等人。成年小鼠大脑的单细胞 DNA 甲基化组和 3D 多组学图谱。自然https://doi.org/10.1038/s41586-023-06805-y (2023)。Yao, Z. et al. A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature https://doi.org/10.1038/s41586-023-06812-z (2023).
Yao, Z. 等人。整个小鼠大脑中细胞类型的高分辨率转录组学和空间图谱。自然https://doi.org/10.1038/s41586-023-06812-z (2023)。Zhang, M. et al. Molecularly defined and spatially resolved cell atlas of the whole mouse brain. Nature https://doi.org/10.1038/s41586-023-06808-9 (2023).
Zhang, M. 等人。整个小鼠大脑的分子定义和空间分辨的细胞图谱。自然https://doi.org/10.1038/s41586-023-06808-9 (2023)。Benevento, M., Hökfelt, T. & Harkany, T. Ontogenetic rules for the molecular diversification of hypothalamic neurons. Nat. Rev. Neurosci. 23, 611–627 (2022).
Benevento, M., Hökfelt, T. & Harkany, T. 下丘脑神经元分子多样化的个体发育规则。Nat. Rev. 神经科学。23, 611–627 (2022 年)。Moffitt, J. R. et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018).
Moffitt, JR 等人。下丘脑视前区域的分子、空间和功能单细胞分析。科学362, eaau5324 (2018)。Usrey, W. M. & Sherman, S. M. (Oxford Univ. Press, 2021).
Usrey, W. M. & Sherman, S. M. (牛津大学出版社, 2021).Sripanidkulchai, K. & Wyss, J. M. Thalamic projections to retrosplenial cortex in the rat. J. Comp. Neurol. 254, 143–165 (1986).
Sripanidkulchai, K. & Wyss, J. M. 大鼠脾后皮层的丘脑突起。J. Comp. 神经。254, 143–165 (1986 年)。Vitalis, T. et al. RORα coordinates thalamic and cortical maturation to instruct barrel cortex development. Cereb. Cortex 28, 3994–4007 (2018).
Vitalis, T. et al. RORα 协调丘脑和皮质成熟以指导桶状皮层发育。大脑。皮质28, 3994–4007 (2018)。Carcea, I. et al. Maturation of cortical circuits requires Semaphorin 7 A. Proc. Natl Acad. Sci. USA 111, 13978–13983 (2014).
Carcea, I. 等人。皮质回路的成熟需要信号素 7 A. Proc. Natl Acad. Sci. USA111, 13978–13983 (2014)。Bosse, A. et al. Identification of the vertebrate Iroquois homeobox gene family with overlapping expression during early development of the nervous system. Mech. Dev. 69, 169–181 (1997).
Bosse, A. 等人。鉴定在神经系统早期发育过程中表达重叠的脊椎动物易洛魁同源框基因家族。机械开发69, 169–181 (1997 年)。Zander, J.-F. et al. Synaptic and vesicular coexistence of VGLUT and VGAT in selected excitatory and inhibitory synapses. J. Neurosci. 30, 7634–7645 (2010).
赞德,J.-F.等。VGLUT 和 VGAT 在选定的兴奋性和抑制性突触中的突触和囊泡共存。J. 神经科学。30, 7634–7645 (2010 年)。Kramer, S. G., Kidd, T., Simpson, J. H. & Goodman, C. S. Switching repulsion to attraction: changing responses to slit during transition in mesoderm migration. Science 292, 737–740 (2001).
Kramer, S. G., Kidd, T., Simpson, J. H. & Goodman, C. S. 将排斥转化为吸引力:在中胚层迁移过程中过渡期间改变对狭缝的反应。科学292, 737–740 (2001)。Hohenester, E., Hussain, S. & Howitt, J. A. Interaction of the guidance molecule Slit with cellular receptors. Biochem. Soc. Trans. 34, 418–421 (2006).
Hohenester, E., Hussain, S. & Howitt, J. A. 引导分子Slit与细胞受体的相互作用。Biochem. Soc. Trans.34, 418–421 (2006 年)。Farghaian, H. et al. Scapinin-induced inhibition of axon elongation is attenuated by phosphorylation and translocation to the cytoplasm. J. Biol. Chem. 286, 19724–19734 (2011).
Farghaian, H. 等人。Scapinin 诱导的轴突伸长抑制通过磷酸化和易位到细胞质而减弱。J. Biol. 化学。286, 19724–19734 (2011)。Miyata, T. et al. Neuron-enriched phosphatase and actin regulator 3 (Phactr3)/nuclear scaffold-associated PP1-inhibiting protein (Scapinin) regulates dendritic morphology via its protein phosphatase 1-binding domain. Biochem. Biophys. Res. Commun. 528, 322–329 (2020).
Miyata, T. 等人。神经元富集磷酸酶和肌动蛋白调节因子 3 (Phactr3)/核支架相关 PP1 抑制蛋白 (Scapinin) 通过其蛋白磷酸酶 1 结合结构域调节树突状形态。生化。生物物理学。Res. Commun.528, 322–329 (2020 年)。Trudeau, L.-E. et al. The multilingual nature of dopamine neurons. Prog. Brain Res. 211, 141–164 (2014).
特鲁多,L.-E.等。多巴胺神经元的多语言性质。程序。大脑研究。211, 141–164 (2014 年)。Bouarab, C., Thompson, B. & Polter, A. M. VTA GABA neurons at the interface of stress and reward. Front. Neural Circuits 13, 78 (2019).
Bouarab, C., Thompson, B. & Polter, A. M. VTA GABA神经元在压力和奖励的界面上。前面。神经回路13, 78 (2019)。Cai, J. & Tong, Q. Anatomy and function of ventral tegmental area glutamate neurons. Front. Neural Circuits 16, 867053 (2022).
Cai, J. & Tong, Q. 腹侧被盖区谷氨酸神经元的解剖和功能。前面。神经回路16, 867053 (2022)。Phillips, R. A. 3rd et al. An atlas of transcriptionally defined cell populations in the rat ventral tegmental area. Cell Rep. 39, 110616 (2022).
菲利普斯,RA 3rd 等人。大鼠腹侧被盖区转录定义的细胞群图谱。Cell Rep.39, 110616 (2022)。Yamaguchi, T., Sheen, W. & Morales, M. Glutamatergic neurons are present in the rat ventral tegmental area. Eur. J. Neurosci. 25, 106–118 (2007).
Yamaguchi, T., Sheen, W. & Morales, M. 谷氨酸能神经元存在于大鼠腹侧被盖区域。Eur. J. 神经科学。25, 106–118 (2007 年)。Yamaguchi, T., Wang, H.-L., Li, X., Ng, T. H. & Morales, M. Mesocorticolimbic glutamatergic pathway. J. Neurosci. 31, 8476–8490 (2011).
Yamaguchi, T., Wang, H.-L., Li, X., Ng, T. H. & Morales, M. 中皮质边缘谷氨酸能途径。J. 神经科学。31, 8476–8490 (2011 年)。Lacar, B. et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun. 7, 11022 (2016).
Lacar, B. 等人。单个神经元的核 RNA-seq 揭示了激活的分子特征。Nat. Commun.7, 11022 (2016 年)。Liu, H. et al. DNA methylation atlas of the mouse brain at single-cell resolution. Nature 598, 120–128 (2021).
Liu, H. et al. 单细胞分辨率下小鼠大脑的 DNA 甲基化图谱。自然598, 120–128 (2021)。Miles, A. et al. zarr-developers/zarr-python: v2.5.0. Zenodo https://doi.org/10.5281/zenodo.4069231 (2020).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Smith, S. J. et al. Single-cell transcriptomic evidence for dense intracortical neuropeptide networks. Elife 8, e47889 (2019).
Tasic, B. et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 19, 335–346 (2016).
González-Blas, C. B. et al. SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks. Nat. Methods 20, 1355–1367 (2023).
Gupta, S., Stamatoyannopoulos, J. A., Bailey, T. L. & Noble, W. S. Quantifying similarity between motifs. Genome Biol. 8, R24 (2007).
Frith, M. C., Li, M. C. & Weng, Z. Cluster-Buster: finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 31, 3666–3668 (2003).
Acknowledgements
We are grateful to M. Nunn for help with management of the project. This work is supported by National Institute of Mental Health grant U19MH114831 to E.M.C. and J.R.E. under the BRAIN Initiative of the National Institutes of Health (NIH) and by National Eye Institute grant R01EY022577 to E.M.C. The Flow Cytometry Core Facility of the Salk Institute (Research Resource Identifier: SCR_014839) is supported by funding from NIH–National Cancer Institute Cancer Center Support Grant P30 014195 and Shared Instrumentation Grant S10-OD023689 (Aria Fusion cell sorter). J.R.E. is an investigator of the Howard Hughes Medical Institute. The 10x scRNA-seq was financed by the grant U19MH114830 to H.Z. under the BRAIN Initiative of the NIH.
Ethics declarations
Competing interests
J.R.E. serves on the scientific advisory board of Zymo Research Inc.
Peer review
Peer review information
Nature thanks Rajeshwar Awatramani and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 The dissection map of source regions across the mouse brain.
The posterior views of dissected slices are shown. The slices correspond to Allen Mouse Common Coordinate Framework (CCF), Reference Atlas, Version 3 (2020)13, level 21 ~ 27 (slice 1), 27 ~ 33 (slice 2), 33 ~ 39 (slice 3), 39 ~ 45 (slice 4), 45 ~ 51 (slice 5), 51 ~ 57 (slice 6), 57 ~ 63 (slice 7), 63 ~ 69 (slice 8), 69 ~ 75 (slice 9), 75 ~ 81 (slice 10), 81 ~ 87 (slice 11), 87 ~ 93 (slice 12), 93 ~ 99 (slice 13), 99 ~ 105 (slice 14), 105 ~ 111 (slice 15), 111 ~ 117 (slice 16), 117 ~ 123 (slice 17), and 123 ~ 129 (slice 18), respectively. Regions dissected from each slice are indicated by dotted lines and are annotated. Allen Brain Reference Atlas, http://www.atlas.brain-map.org, © 2017 Allen Institute for Brain Science.
Extended Data Fig. 2 Quality control workflow and region group assignment.
a, b, Joint t-SNE of Epi-Retro-Seq cells (n = 56,843) and unbiased snmC-seq cells (n = 310,605) after basic QC (Methods, QC Step 1) colored by the predicted outliers (a) or the total number of reads normalized per sequencing plate (b). c, d, Joint t-SNE of Epi-Retro-Seq cells (n = 48,032) and unbiased snmC-seq cells (n = 301,626) after removing outlier clusters (Methods, QC Step 2) colored by neuronal vs. non-neuronal cells (c) or their assigned L1 type (d). e, the on-target vs. off-target fold enrichment (x-axis) and -log10 FDR (y-axis) of IT (n = 186, top) or ET (n = 100, bottom) FANS experiments. The size of the circle is proportional to the number of neurons captured in the experiment. f, The overlap scores between 115 snmC-seq dissections and 87 scRNA-seq dissections. Each region group is colored differently on the x and y axes and squared in the heatmap. g, Joint t-SNE of Epi-Retro-Seq (n = 35,743), snmC-seq (n = 266,740), and scRNA-seq (n = 2,434,472) neurons colored by region groups. (f) and (g) share the same color palette.
Extended Data Fig. 3 Quantification of target discriminability for all 926 target pairs from all source regions.
a, Comparisons of AUROCs from models trained with gene features (x-axis) or 100 kb bin features (y-axis) using posterior mCH level (top) or mCH principal components (bottom) when splitting the training and testing data according to biological replicates (left) or computational replicates (right). b, Comparisons of AUROCs from models trained with mCH principal components (x-axis) or posterior mCH level (y-axis) using gene (top) or 100 kb bin (bottom) features when split the training and testing data according to biological replicates (left) or computational replicates (right). c, Comparisons of AUROCs from models when splitting the training and testing data according to biological replicates (x-axis) or computational replicates (y-axis) using gene (top) or 100 kb bin (bottom) features and posterior mCH level (left) or mCH principal components (right). For a-c, each dot represents a pairwise comparison between the neurons projecting from the same source to two different targets. The plots involving biological replicates have 516 data points each while the others have 926 data points each. Pearson Correlation Coefficient (PCC, r) and P value (permutation test) are labeled in each panel. d, The AUROC between neurons projecting from each of the 30 sources to all possible pairs of targets that have been profiled for the source. STR and CBX are not included since we only profiled one target for these sources.
Extended Data Fig. 4 Co-clustering of Epi-Retro-Seq, unbiased snmC-seq and scRNA-seq data for 12 brain regions.
For each brain region group, the joint t-SNE colored by data modality (left) and the proportion of cells from each snmC-seq L4 cluster (middle, column) or scRNA-seq L3 cluster (right, column) within each co-cluster (middle and right, row). Numbers of co-clusters in the rows are sometimes different for middle and right columns because only the co-clusters (rows) with more than one value > 10% across the columns are shown. See Methods for further details.
Extended Data Fig. 5 Projection-enriched cell clusters and their neurotransmitter usage for all brain regions.
Joint clustering analysis of Epi-Retro-Seq, unbiased snmC-seq and scRNA-seq was performed on each of the major brain region groups, including CTX, RHP, PIR, HB, MOB + AON, AMY, TH, HIP, MB, HY, and PAL (STR not included because there is only one target), to characterize neuronal cell clusters that were enriched for Epi-Retro-Seq projections. The normalized proportion of each projection in each cluster was visualized in the heatmaps (left) for each of the brain region groups. “*” denotes FDR < 0.01 for testing if the proportion of cells in a cluster among those projecting to a target is greater than the proportion of cells in this cluster among those projecting to all other targets (one-sided Fisher exact test; Benjamini-Hochberg procedure). “X” denotes FDR < 0.01 for testing if the proportion of cells in a cluster among those projecting to a target is different between male and female samples (two-sided Fisher exact test; Benjamini-Hochberg procedure). In addition, the expression levels of 10 marker genes for neurotransmitter usage in each cluster are visualized in the heatmap (right) for each brain region group. These genes included Slc17a7 (Vglut1), Slc17a6 (Vglut2), and Slc17a8 (Vglut3) for glutamatergic neurons, Slc32a1 (Vgat) for GABAergic neurons, Slc6a2 (Net) for noradrenergic neurons, Slc6a3 (Dat) for dopaminergic neurons, Slc6a4 (Sert) for serotonergic neurons, Slc6a5 (Glyt2) for glycinergic neurons, Slc18a3 (Vacht) for cholinergic neurons, and histidine decarboxylase (Hdc) for histaminergic neurons.
Extended Data Fig. 6 Joint clustering and annotation of MERFISH and scRNA-seq data.
a, b, Joint t-SNE of scRNA-seq neurons (n = 2,619,158, left) and MERFISH neurons (n = 329,282, right) colored by cell subclass (a) or region group (b). The labels for scRNA-seq cells are based on Yao et al.18 and the labels for MERFISH cells are predicted through integration. c, d, The MERFISH slices colored by cell subclass (c) or region group (d). Scale bars represent 15 mm.
Extended Data Fig. 7 Integration and comparisons of hypothalamic Epi-Retro-Seq, Act-seq, and scRNAseq data.
a, b, Joint t-SNE of Act-Seq neurons7 (n = 78,476, a) and scRNA-seq neurons (n = 148,840, b). In (a), all Act-Seq neurons (left) or only VMHvl neurons (right) are colored by Act-Seq neuron cluster (left) or VMH cluster (right). In (b), all scRNA-seq neurons (left) or only neurons in projection associated clusters (right) are colored by the co-cluster label. c, d, The proportion of neurons from each of the Act-Seq VMHvl clusters (c) or all neuron clusters (d) classified as neurons of each co-cluster. Only the co-clusters with value > 0.1 in at least one Act-Seq cluster are shown. The projection-associated co-clusters are labeled in red. e, f, Proportion of Fos+ “behavior activated” cells (left) or average Fos expression (right) of each VMHvl cluster (e) or each co-cluster (f) in control and different behavior experiments. Only the co-clusters labeled red in d are shown in (f). *, **, and *** represent FDR < 0.1, 0.01, and 0.001, respectively.
Extended Data Fig. 8 Comparison of thalamic Retro-Seq and Epi-Retro-Seq data.
a, t-SNEs for visualization of the Retro-Seq data from thalamic neurons projecting to prefrontal, motor, somatosensory, auditory, and visual cortices8 that were mapped onto the joint-clustering analysis of Epi-Retro-Seq, unbiased snmC-seq and scRNA-seq in TH. b, The t-SNEs for visualization of the Epi-Retro-Seq data for thalamic neurons projecting to 12 different targets that were mapped onto the same t-SNE space. c, The overlap score and cosine distance were calculated for each pairwise comparison of Retro-Seq and Epi-Retro-Seq projections and were visualized in the heatmaps, respectively.
Extended Data Fig. 9 The neurotransmitter usage of VTA projection neurons.
a, Joint t-SNE of Epi-Retro-Seq, unbiased snmC-seq and scRNA-seq of VTA neurons colored by the gene expression levels (red) and gene-body mCH levels (purple) for Vglut2 (left), Gad2 (middle), and Th (right), marker genes for glutamatergic, GABAergic, and dopaminergic neurons, respectively. b, The distribution of VTA neurons projecting to each of the 16 targets on the same t-SNE. c-e, The gene-body mCH levels of Th versus Vglut2 (c), Gad2 versus Vglut2 (d), and Gad2 versus Th (e) for VTA neurons projecting to each of the 16 targets, are visualized in scatter plots. Note that, because low mCH levels indicate high gene expression, the axes in c-e are plotted as the reciprocal mCH values (1/gene body mCH), so low mCH is plotted to the right/up and high to the left/down.
Supplementary information
Supplementary Table 1
Epi-retro-seq injection information.
Supplementary Table 2
Epi-retro-seq cell metadata.
Supplementary Table 3
Number of cells that passed QC for source–target combinations.
Supplementary Table 4
AUROC scores for target pairs.
Supplementary Table 5
Gene categories for AUROC analyses.
Supplementary Table 6
FDR for AUROC comparisons between gene categories.
Supplementary Table 7
Summary of projection-enriched clusters in HY and TH.
Supplementary Table 8
Comparisons of injection coordinates to retrogradely label thalamic projection neurons for epi-retro-seq versus retro-seq.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, J., Zhang, Z., Wu, M. et al. Brain-wide correspondence of neuronal epigenomics and distant projections. Nature 624, 355–365 (2023). https://doi.org/10.1038/s41586-023-06823-w
This article is cited by
-
Parallel labeled-line organization of sympathetic outflow for selective organ regulation in mice
Nature Communications (2024)
-
The single-cell opioid responses in the context of HIV (SCORCH) consortium
Molecular Psychiatry (2024)
-
Cellular atlases of the entire mouse brain
Nature (2023)