
Abstract 抽象的
Ferroptosis is a non-apoptotic cell death mechanism characterized by iron-dependent membrane lipid peroxidation. Here, we review what is known about the cellular mechanisms mediating the execution and regulation of ferroptosis. We first consider how the accumulation of membrane lipid peroxides leads to the execution of ferroptosis by altering ion transport across the plasma membrane. We then discuss how metabolites and enzymes that are distributed in different compartments and organelles throughout the cell can regulate sensitivity to ferroptosis by impinging upon iron, lipid and redox metabolism. Indeed, metabolic pathways that reside in the mitochondria, endoplasmic reticulum, lipid droplets, peroxisomes and other organelles all contribute to the regulation of ferroptosis sensitivity. We note how the regulation of ferroptosis sensitivity by these different organelles and pathways seems to vary between different cells and death-inducing conditions. We also highlight transcriptional master regulators that integrate the functions of different pathways and organelles to modulate ferroptosis sensitivity globally. Throughout this Review, we highlight open questions and areas in which progress is needed to better understand the cell biology of ferroptosis.
铁死亡是一种非凋亡细胞死亡机制,其特征是铁依赖性膜脂质过氧化。在这里,我们回顾了关于介导铁死亡的执行和调节的细胞机制的已知信息。我们首先考虑膜脂质过氧化物的积累如何通过改变跨质膜的离子传输来导致铁死亡的执行。然后我们讨论分布在整个细胞不同区室和细胞器中的代谢物和酶如何通过影响铁、脂质和氧化还原代谢来调节对铁死亡的敏感性。事实上,线粒体、内质网、脂滴、过氧化物酶体和其他细胞器中的代谢途径都有助于铁死亡敏感性的调节。我们注意到这些不同的细胞器和途径对铁死亡敏感性的调节似乎在不同的细胞和死亡诱导条件之间存在差异。我们还重点介绍了转录主调控因子,它们整合了不同途径和细胞器的功能,以全局调节铁死亡敏感性。在这篇综述中,我们强调了一些悬而未决的问题和需要取得进展的领域,以更好地了解铁死亡的细胞生物学。
Similar content being viewed by others
其他人正在查看类似内容

Introduction 介绍
Ferroptosis is a non-apoptotic cell death mechanism that requires the redox-active metal iron1,2. Free intracellular iron or iron-containing enzymes react with oxygen and polyunsaturated fatty acid (PUFA)-containing lipids to generate high levels of membrane lipid peroxides — the defining feature of ferroptosis in relation to other forms of cell death3 (Box 1). These membrane lipid peroxides can be lethal to the cell when they accumulate at high levels. Any cell in the biosphere containing iron, oxygen and PUFA-containing lipids may therefore be at risk of undergoing ferroptosis. For this reason, cells have evolved powerful enzyme-catalysed mechanisms to defend against oxidative membrane damage and the onset of ferroptosis4,5 (Fig. 1a). Ferroptosis is a physiological process with a role in homeostasis, namely tumour suppression6,7. Ferroptosis may also be activated in acute and chronic disease conditions8. Accordingly, there is considerable interest in understanding the nature and regulation of this mechanism.
铁死亡是一种非凋亡性细胞死亡机制,需要氧化还原活性金属铁1 , 2 。细胞内游离铁或含铁酶与氧和含多不饱和脂肪酸(PUFA) 的脂质发生反应,产生高水平的膜脂过氧化物——铁死亡与其他形式的细胞死亡相关的决定性特征3 (框1 )。这些膜脂过氧化物在高水平积累时可能对细胞致命。因此,生物圈中任何含有铁、氧和含有 PUFA 的脂质的细胞都可能面临铁死亡的风险。因此,细胞进化出强大的酶催化机制来防御氧化膜损伤和铁死亡的发生4 , 5 (图1a )。铁死亡是一种具有稳态作用的生理过程,即肿瘤抑制6 , 7 。铁死亡也可能在急性和慢性疾病中被激活8 。因此,人们对了解这种机制的性质和调节非常感兴趣。
图1:铁死亡的一般机制。

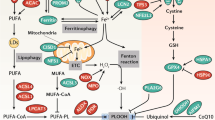
a, Ferroptosis execution can be distinguished from the regulation of ferroptosis sensitivity. Ferroptosis execution involves membrane lipid peroxidation, leading to plasma membrane rupture and cell death. The glutathione (GSH)–glutathione peroxidase 4 (GPX4) and NADPH–ferroptosis suppressor protein 1 (FSP1) systems limit membrane lipid peroxidation and inhibit ferroptosis. Ferroptosis sensitivity is dictated by the propensity of the cell to accumulate high levels of membrane lipid peroxides. Whether a cell will accumulate high levels of lipid peroxides relates to the amount of iron and oxidizable lipids in the cell, and to the status of the cellular oxidant-producing and antioxidant systems. The regulation of ferroptosis sensitivity likely encompasses hundreds of distinct metabolites, enzymes and other biomolecules that impinge upon the membrane lipid peroxidation and membrane-rupturing mechanisms. b, Plasma membrane rupture is the key event that results in cell death during ferroptosis. It involves the accumulation of phospholipid hydroperoxides, which may be generated enzymatically (for example, via lipoxygenases) and non-enzymatically through radical-mediated reactions. Phospholipid peroxidation has several consequences for the cell, including altered ion fluxes (for example, increased Piezo-mediated Ca2+ uptake, increased transient receptor potential (TRP)-mediated Ca2+ and Na+ uptake, and reduced Na+/K+ ATPase-mediated Na+ export and K+ uptake), water ingress and biophysical effects on the membrane. Cell swelling and increased membrane stiffness can further activate ion channels (for example, Piezo and TRP) in a feedforward manner, further enhancing ion fluxes and accelerating plasma membrane rupture. c, Important ferroptosis defence mechanisms localize to the plasma membrane. The system xc– antiporter imports cystine in exchange for glutamate. In the cytosol, cystine is rapidly reduced to cysteine. Cysteine can be used to synthesize (reduced) GSH. GSH is the cofactor for GPX4, an enzyme that can convert toxic phospholipid peroxides (L-OOH) into benign lipid alcohols (L-OH). Parallel to GPX4, the oxidoreductase FSP1 uses NAD(P)H to regenerate the reduced form of radical-trapping antioxidants (for example, coenzyme Q10 (CoQ) or vitamin K), which in turn terminate the lipid peroxidation process by donating electrons to phospholipid peroxyl radicals (LOO•). Receptor-mediated endocytosis influences ferroptosis sensitivity. For example, the uptake of iron in complex with transferrin by the transferrin receptor enhances ferroptosis sensitivity while uptake of the selenium-rich protein SEPP1 by its cognate receptor LDL receptor-related protein 8 (LRP8) suppresses ferroptosis sensitivity. Iron, and presumably selenium in the form of selenocysteine, are subsequently released from the lysosome. Iron can react with soluble and lipid peroxides to generate hydroxyl and lipid alkoxyl radicals that promote lipid peroxidation. Cysteine and selenium released from the lysosome can be used to synthesize GSH and the selenoprotein GPX4, respectively. ACSL4, acyl-CoA synthetase long-chain family member 4; CHMP, charged multivesicular body protein; CoA, coenzyme A; GR, GSH reductase; GSSG, oxidized GSH; PKCβII, protein kinase Cβ; PUFA, polyunsaturated fatty acid.
a ,铁死亡的执行可以与铁死亡敏感性的调节区分开来。铁死亡执行涉及膜脂过氧化,导致质膜破裂和细胞死亡。谷胱甘肽 (GSH)-谷胱甘肽过氧化物酶 4 (GPX4) 和 NADPH-铁死亡抑制蛋白 1 (FSP1) 系统限制膜脂过氧化并抑制铁死亡。铁死亡敏感性由细胞积累高水平膜脂过氧化物的倾向决定。细胞是否会积累高水平的脂质过氧化物与细胞中铁和可氧化脂质的量以及细胞产生氧化剂和抗氧化系统的状态有关。铁死亡敏感性的调节可能涉及数百种不同的代谢物、酶和其他影响膜脂质过氧化和膜破裂机制的生物分子。 b ,质膜破裂是铁死亡过程中导致细胞死亡的关键事件。它涉及磷脂氢过氧化物的积累,其可以通过酶促(例如,通过脂氧合酶)和非酶促通过自由基介导的反应产生。磷脂过氧化对细胞有多种影响,包括改变离子通量(例如,增加压电介导的 Ca 2+吸收、增加瞬时受体电位 (TRP) 介导的 Ca 2+和 Na +吸收以及减少 Na + /K + ATP 酶介导的 Na +输出和 K +吸收)、水进入和对膜的生物物理影响。 细胞肿胀和膜硬度增加可以前馈方式进一步激活离子通道(例如,Piezo 和 TRP),进一步增强离子通量并加速质膜破裂。 c ,重要的铁死亡防御机制定位于质膜。系统 x c –逆向转运蛋白导入胱氨酸以换取谷氨酸。在细胞质中,胱氨酸迅速还原为半胱氨酸。半胱氨酸可用于合成(还原型)GSH。 GSH 是 GPX4 的辅助因子,GPX4 是一种可以将有毒的磷脂过氧化物 (L-OOH) 转化为良性脂质醇 (L-OH) 的酶。与 GPX4 类似,氧化还原酶 FSP1 使用 NAD(P)H 再生还原形式的自由基捕获抗氧化剂(例如辅酶 Q10 (CoQ) 或维生素 K),从而通过向磷脂提供电子来终止脂质过氧化过程过氧自由基(LOO • )。受体介导的内吞作用影响铁死亡敏感性。例如,转铁蛋白受体对与转铁蛋白复合物的铁的摄取增强了铁死亡的敏感性,而富硒蛋白SEPP1通过其同源受体LDL受体相关蛋白8(LRP8)的摄取则抑制了铁死亡的敏感性。随后,铁以及可能以硒代半胱氨酸形式存在的硒从溶酶体中释放出来。铁可以与可溶性过氧化物和脂质过氧化物反应,生成羟基和脂质烷氧基自由基,促进脂质过氧化。从溶酶体释放的半胱氨酸和硒可分别用于合成谷胱甘肽和硒蛋白GPX4。 ACSL4,酰基辅酶A合成酶长链家族成员4; CHMP,带电多泡体蛋白; CoA、辅酶A; GR、谷胱甘肽还原酶; GSSG,氧化谷胱甘肽; PKCβII,蛋白激酶Cβ; PUFA,多不饱和脂肪酸。
We can distinguish the execution of ferroptosis from mechanisms that regulate ferroptosis sensitivity. The execution of ferroptosis involves membrane lipid peroxidation, the aberrant movement of ions across the plasma membrane, cell swelling and plasma membrane rupture9. Cellular sensitivity to ferroptosis — the likelihood that a given stimulus will cause plasma membrane rupture through the ferroptosis mechanism — is regulated positively and negatively by molecules and pathways that control lipid metabolism, iron homeostasis, redox regulation and related processes. Accordingly, sensitivity to ferroptosis is governed by hundreds of enzymes, reactions and molecules in the cell10. These species are distributed throughout the plasma membrane and cytosol, as well as in different organelles. Depending on the cell type and the ferroptosis-inducing condition, different metabolites, enzymes and organelles appear more important than others for ferroptosis execution and the regulation of ferroptosis sensitivity10. Understanding this context-specific regulation of ferroptosis sensitivity presents an interesting challenge.
我们可以将铁死亡的执行与调节铁死亡敏感性的机制区分开来。铁死亡的发生涉及膜脂过氧化、离子穿过质膜的异常运动、细胞肿胀和质膜破裂9 。细胞对铁死亡的敏感性(特定刺激通过铁死亡机制导致质膜破裂的可能性)受到控制脂质代谢、铁稳态、氧化还原调节和相关过程的分子和途径的正向和负向调节。因此,对铁死亡的敏感性由细胞10中的数百种酶、反应和分子控制。这些物种分布在整个质膜和细胞质以及不同的细胞器中。根据细胞类型和铁死亡诱导条件,不同的代谢物、酶和细胞器对于铁死亡的执行和铁死亡敏感性的调节似乎比其他代谢物更重要10 。了解铁死亡敏感性的这种特定环境调节提出了一个有趣的挑战。
In this Review, we examine ferroptosis mechanisms at the cellular level. We first describe the events that lead to lethal plasma membrane rupture. We then discuss how different enzymes and molecules regulate ferroptosis sensitivity from different locations in the cell. Finally, we point out examples of how these processes may be integrated between different regions of the cell to execute or prevent ferroptosis. Throughout this Review, we highlight unanswered questions that exist in connection with the cell biology of ferroptosis.
在这篇综述中,我们研究了细胞水平的铁死亡机制。我们首先描述导致致命质膜破裂的事件。然后我们讨论不同的酶和分子如何从细胞的不同位置调节铁死亡敏感性。最后,我们指出了如何在细胞的不同区域之间整合这些过程以执行或预防铁死亡的示例。在这篇综述中,我们强调了与铁死亡的细胞生物学有关的未解答的问题。
The execution and regulation of ferroptosis at the plasma membrane
Plasma membrane rupture is the terminal event in many forms of cell death. For some forms of non-apoptotic cell death, such as necroptosis and pyroptosis, this terminal process involves permeabilization of the plasma membrane by pore-forming proteins such as mixed lineage kinase domain-like pseudokinase (MLKL), gasdermin D and ninjurin 1 (refs. 11,12). Ninjurin 1 may facilitate the execution of ferroptosis in some but not all cells9,13. Otherwise, there is little evidence that the execution of ferroptosis requires a specific pore-forming protein. Rather, ferroptosis execution and regulation involve a distinct set of lipid-centric mechanisms that govern plasma membrane integrity.
Ferroptosis execution at the plasma membrane
The execution of ferroptosis involves the peroxidation of PUFA-containing phospholipids and the formation of lipid hydroperoxides14,15. Once formed, lipid hydroperoxides can react with iron to generate highly reactive lipid radicals and become further modified or truncated in ways that likely contribute to the execution of ferroptosis16. Lipid hydroperoxides may be synthesized by lipoxygenase enzymes17 or formed through chemical reactions between PUFAs, soluble reactive oxygen species (ROS) and iron10. It is controversial which mechanism is most important for ferroptosis and it seems likely that both mechanisms are relevant in one cell type or another17,18,19,20. Within the membrane, dozens of different PUFA-containing phospholipid species can be peroxidized to contribute to the execution of ferroptosis, with PUFA-phosphatidylethanolamines being especially important in many cells14,21,22,23,24,25,26. When cells are exposed to ferroptosis-inducing conditions, lipid peroxides seem to initially accumulate within the endoplasmic reticulum (ER) and other organelles and subsequently at the plasma membrane prior to membrane rupture27,28,29,30 (Box 2).
Events downstream of plasma membrane lipid peroxidation that promote ferroptosis execution have recently been clarified (Fig. 1b). Increasing lipid peroxidation raises membrane tension, which in turn activates Piezo1 and transient receptor potential (TRP) mechanosensitive ion channels9. Channel opening leads to Ca2+ and Na+ influx and K+ efflux. These ion fluxes are enhanced by the simultaneous inactivation of the plasma membrane Na+/K+ ATPase, possibly due to altered interactions between this protein and peroxidized plasma membrane phospholipids9. Collectively, these changes lead to a loss of ionic homeostasis and osmotic cell swelling that eventually result in plasma membrane rupture. Plasma membrane rupture can be transiently delayed by polyethylene glycol molecules that block membrane nanopores to prevent the flow of ions and water across the plasma membrane31,32. However, polyethylene glycol molecules only delay the onset of ferroptosis and do not completely prevent plasma membrane rupture over longer periods of time31. It seems likely that, as more lipids become peroxidized, this disrupts the biophysical properties of the plasma membrane33 so severely that plasma membrane rupture becomes inevitable.
最近已经阐明了质膜脂质过氧化下游促进铁死亡执行的事件(图1b )。脂质过氧化的增加会提高膜张力,进而激活 Piezo1 和瞬时受体电位 (TRP) 机械敏感离子通道9 。通道开放导致Ca 2+和Na +流入以及K +流出。这些离子通量通过质膜 Na + /K + ATP 酶的同时失活而增强,这可能是由于该蛋白质与过氧化质膜磷脂之间的相互作用发生改变9 。总的来说,这些变化导致离子稳态丧失和渗透细胞肿胀,最终导致质膜破裂。质膜破裂可以通过聚乙二醇分子暂时延迟,聚乙二醇分子阻塞膜纳米孔以防止离子和水流过质膜31、32 。然而,聚乙二醇分子只能延迟铁死亡的发生,并不能在较长时间内完全防止质膜破裂31 。似乎有可能,随着更多的脂质被过氧化,这会严重破坏质膜33的生物物理性质,以致质膜破裂变得不可避免。
Suppression of membrane lipid peroxidation
抑制膜脂过氧化
Ferroptosis is normally inhibited by the continuous activity of coupled enzyme-metabolite systems that prevent the accumulation of membrane lipid peroxides to toxic levels. These systems are, in fact, the targets of small molecules that helped elucidate the ferroptosis mechanism in the first place (see Box 2 for a description of common ferroptosis inducers and inhibitors). The enzyme glutathione peroxidase 4 (GPX4) reduces potentially toxic lipid hydroperoxides to less dangerous lipid alcohols34,35. GPX4 appears to be the most critical enzyme for preventing lipid hydroperoxide accumulation in most cells35,36 (Fig. 1c). Several isoforms of GPX4 exist, including those designated as cytosolic, mitochondrial and nuclear. Cytosolic GPX4 seems the most essential for preventing ferroptosis, although mitochondrial GPX4 may also help inhibit ferroptosis in some cases37,38,39. Cytosolic GPX4 does not contain an obvious plasma membrane-targeting element. However, modelling studies indicate that a patch of positively charged lysine and arginine residues may facilitate the electrostatic interactions with phospholipids that help keep GPX4 in the vicinity of the plasma membrane40.
铁死亡通常受到偶联酶代谢系统的持续活性的抑制,该系统可防止膜脂过氧化物累积至有毒水平。事实上,这些系统是小分子的靶标,这些小分子首先帮助阐明了铁死亡机制(参见方框2 ,了解常见铁死亡诱导剂和抑制剂的描述)。谷胱甘肽过氧化物酶 4 (GPX4) 将潜在有毒的脂质氢过氧化物还原为危险性较低的脂质醇34 、 35 。 GPX4 似乎是防止大多数细胞中脂质氢过氧化物积累的最关键酶35 、 36 (图1c )。 GPX4 存在多种亚型,包括细胞质亚型、线粒体亚型和核亚型。胞质 GPX4 似乎对于预防铁死亡最重要,尽管线粒体 GPX4 在某些情况下也可能有助于抑制铁死亡37 , 38 , 39 。胞质 GPX4 不包含明显的质膜靶向元件。然而,模型研究表明,一片带正电的赖氨酸和精氨酸残基可能促进与磷脂的静电相互作用,从而有助于将 GPX4 保持在质膜附近40 。
Ferroptosis suppressor protein 1 (FSP1; also known as AIFM2) inhibits plasma membrane lipid peroxidation in parallel to GPX4 (Fig. 1c). FSP1 is an NAD(P)H- and FAD-dependent oxidoreductase that reduces coenzyme Q10 (CoQ)41,42 and vitamin K43. The reduced forms of CoQ and vitamin K act as lipophilic radical-trapping antioxidants (RTAs) that can terminate lipid peroxidation chain reactions. FSP1 localizes specifically to the plasma membrane and to lipid droplets41,42. During translation, the N-terminal initiator methionine of FSP1 is removed and the 14-carbon fatty acid myristate is covalently attached to the subsequent glycine residue41,42. The irreversibly conjugated myristate anchors FSP1 to the plasma membrane and is necessary for its anti-ferroptotic activity41,42. Targeting FSP1 selectively to the plasma membrane is sufficient to suppress ferroptosis, implying that this is the key site where endogenous RTAs function to suppress ferroptosis41. Co-immunoprecipitation and biochemistry data support the possibility that human FSP1 exists as a dimer44, but whether this is the functional state of FSP1 at the membrane is unknown. In addition to CoQ and vitamin K, other endogenous metabolites can serve as RTA inhibitors of ferroptosis, including tetrahydrobiopterin23,45, vitamin E and vitamin A46. The relative contribution of these different metabolic ferroptosis suppressors appears to vary between cell types.
铁死亡抑制蛋白1(FSP1;也称为AIFM2)与GPX4同时抑制质膜脂质过氧化(图1c )。 FSP1 是一种 NAD(P)H 和 FAD 依赖性氧化还原酶,可还原辅酶 Q10 (CoQ) 41 、 42和维生素 K 43 。还原形式的 CoQ 和维生素 K 作为亲脂性自由基捕获抗氧化剂 (RTA),可以终止脂质过氧化链反应。 FSP1特异性定位于质膜和脂滴41、42 。在翻译过程中,FSP1的N端起始子甲硫氨酸被去除,并且14碳脂肪酸肉豆蔻酸酯共价连接至随后的甘氨酸残基41、42 。不可逆结合的肉豆蔻酸酯将 FSP1 锚定在质膜上,并且是其抗铁死亡活性所必需的41 , 42 。将 FSP1 选择性地靶向质膜足以抑制铁死亡,这意味着这是内源性 RTA 发挥抑制铁死亡作用的关键位点41 。免疫共沉淀和生物化学数据支持人类 FSP1 以二聚体形式存在的可能性44 ,但这是否是 FSP1 在膜上的功能状态尚不清楚。除了 CoQ 和维生素 K 之外,其他内源性代谢物也可以作为铁死亡的 RTA 抑制剂,包括四氢生物蝶呤23 、 45 、维生素 E 和维生素 A 46 。这些不同的代谢性铁死亡抑制剂的相对贡献似乎因细胞类型而异。
Plasma membrane repair as an anti-ferroptotic mechanism
质膜修复作为抗铁死亡机制
Plasma membrane repair processes oppose the terminal execution of ferroptosis. As noted above, lipid peroxidation can trigger an increase in intracellular Ca2+ levels31. Increased intracellular Ca2+ acts as a signal to recruit charged multivesicular body protein 5 (CHMP5) and CHMP6 — the components of the endosomal sorting complexes required for transport (ESCRT)-III — to the plasma membrane47, where they participate in local membrane repair. Indeed, genetic silencing of CHMP5, CHMP6 or CHMP4B can enhance ferroptosis sensitivity to some degree31,47 (Fig. 1b). However, membrane repair mediated by CHMP4B, CHMP5 and CHMP6 can be overwhelmed by high levels of lipid peroxidation31,47. Of note, the ESCRT-III complex can also limit protein pore-dependent membrane damage during necroptosis and pyroptosis48,49 and is therefore a negative regulator of several forms of non-apoptotic cell death.
质膜修复过程反对铁死亡的最终执行。如上所述,脂质过氧化可以引发细胞内Ca 2+水平的增加31 。细胞内 Ca 2+增加充当信号,将带电多泡体蛋白 5 (CHMP5) 和 CHMP6(运输 (ESCRT)-III 所需的内体分选复合物的成分)招募到质膜47 ,在那里它们参与局部膜维修。事实上, CHMP5 、 CHMP6或CHMP4B的基因沉默可以在一定程度上增强铁死亡敏感性31、47 (图1b )。然而,CHMP4B、CHMP5 和 CHMP6 介导的膜修复可能会被高水平的脂质过氧化作用所淹没31 、 47 。值得注意的是,ESCRT-III 复合物还可以限制坏死性凋亡和焦亡期间蛋白质孔依赖性膜损伤48 、 49 ,因此是几种形式的非凋亡细胞死亡的负调节因子。
Ferroptosis regulation by plasma membrane transporters and enzymes
质膜转运蛋白和酶对铁死亡的调节
Integral plasma membrane proteins can contribute to the execution and regulation of ferroptosis in many ways, including through the generation of ROS1,50,51, and to the mediation of iron or metabolite trafficking52,53,54,55,56,57. Here, we focus on proteins that are involved in the import of cysteine and selenium, two metabolites that contribute essentially to the negative regulation of ferroptosis (see also next section) (Fig. 1c).
完整的质膜蛋白可以通过多种方式促进铁死亡的执行和调节,包括通过ROS的产生1,50,51 ,以及介导铁或代谢物运输52,53,54,55,56,57 。在这里,我们重点关注参与半胱氨酸和硒输入的蛋白质,这两种代谢物基本上有助于铁死亡的负调节(另见下一节)(图1c )。
Cysteine is required to synthesize sulfur-containing anti-ferroptotic molecules, including the GPX4 cofactor (reduced) glutathione (GSH)58. The cysteine disulfide, cystine, can be imported by the cell surface system xc– cystine–glutamate antiporter. Cysteine can also be acquired indirectly via endocytosis of extracellular cysteine-rich proteins, such as albumin, and their subsequent lysosomal catabolism59. Extracellular GSH can also directly fuel de novo intracellular GSH synthesis via the γ-glutamyl cycle. Plasma membrane-localized γ-glutamyltransferase 1 (GGT1) cleaves the usual γ-glutamyl bond that links glutamate to cysteine in the GSH tripeptide. The resulting cysteinylglycine dipeptide can then be imported and further catabolized by dipeptidases, including carnosine dipeptidase 2 (CNDP2), to release cysteine for use in GSH synthesis60. Some brain cancer cells overcome the need for system xc–-mediated cystine import via GGT1-mediated catabolism of extracellular GSH61. Moreover, in a rare kidney cancer (chromophobe renal cell carcinoma), the absence of GGT1 function renders these cells exquisitely dependent upon extracellular cystine for survival relative to normal tissues62. This helps illustrate how differences in protein expression between cells can result in unique sensitivities to pro-ferroptotic stimuli such as cystine deprivation.
合成含硫抗铁死亡分子需要半胱氨酸,包括 GPX4 辅因子(还原型)谷胱甘肽 (GSH) 58 。半胱氨酸二硫化物胱氨酸可以通过细胞表面系统 x c –胱氨酸 – 谷氨酸逆向转运蛋白输入。半胱氨酸也可以通过细胞外富含半胱氨酸的蛋白质(例如白蛋白)的内吞作用及其随后的溶酶体分解代谢来间接获得59 。细胞外谷胱甘肽还可以通过γ-谷氨酰循环直接促进细胞内谷胱甘肽的从头合成。质膜定位的 γ-谷氨酰转移酶 1 (GGT1) 可裂解 GSH 三肽中连接谷氨酸和半胱氨酸的常见 γ-谷氨酰键。然后,所得半胱氨酰甘氨酸二肽可被输入并进一步被二肽酶(包括肌肽二肽酶 2 (CNDP2))分解代谢,以释放半胱氨酸用于 GSH 合成60 。一些脑癌细胞通过 GGT1 介导的细胞外 GSH 61分解代谢克服了系统 x c介导的胱氨酸输入的需要。此外,在一种罕见的肾癌(嫌色肾细胞癌)中,GGT1 功能的缺失使得这些细胞相对于正常组织而言完全依赖于细胞外胱氨酸的生存62 。这有助于说明细胞之间蛋白质表达的差异如何导致对促铁死亡刺激(例如胱氨酸剥夺)的独特敏感性。
Selenium is found within the active-site selenocysteine of GPX4 and a small number of other enzymes. In cancer cells, high levels of GPX4 translation promote ferroptosis resistance but, under conditions of low selenium, this GPX4 synthesis is inhibited by ribosome stalling and collisions, which causes early GPX4 translation termination (see also next section)63. Selenium can enter the cell by endocytosis of the liver-secreted, selenium-containing carrier protein selenoprotein P, which is bound and endocytosed by cell surface receptors such as the LDL receptor-related protein 8 (LRP8)63,64,65. In triple-negative breast cancer cells, LRP8 knockout sensitizes cells to ferroptosis inducers63. In MYCN-amplified neuroblastoma cells, loss of LRP8 alone is sufficient to trigger ferroptosis, likely due to coincident low expression of system xc– subunits65. Selenium can also enter the cell via the conversion of inorganic selenium (that is, selenite) to volatile selenide through a process that involves system xc–-dependent cystine uptake, which provides thiols that can reduce extracellular inorganic selenium to selenide, which is then presumed to diffuse across the plasma membrane66. How cystine and selenium uptake and metabolism are coordinated to ensure proper GPX4 function is an interesting open question.
硒存在于 GPX4 和少数其他酶的活性位点硒代半胱氨酸中。在癌细胞中,高水平的 GPX4 翻译会促进铁死亡抵抗,但在低硒条件下,这种 GPX4 合成会受到核糖体停滞和碰撞的抑制,从而导致 GPX4 翻译早期终止(另见下一节) 63 。硒可以通过肝脏分泌的含硒载体蛋白硒蛋白P 的内吞作用进入细胞,该蛋白与细胞表面受体如 LDL 受体相关蛋白 8 (LRP8) 63 , 64 , 65结合并被内吞。在三阴性乳腺癌细胞中,LRP8 敲除使细胞对铁死亡诱导剂敏感63 。在 MYCN 扩增的神经母细胞瘤细胞中,仅 LRP8 缺失就足以引发铁死亡,这可能是由于系统 x c –亚基的同时低表达所致65 。硒还可以通过无机硒(即亚硒酸盐)转化为挥发性硒化物进入细胞,该过程涉及系统 x c -依赖的胱氨酸摄取,该过程提供硫醇,可将细胞外无机硒还原为硒化物,然后将其转化为硒化物。推测扩散穿过质膜66 。胱氨酸和硒的摄取和代谢如何协调以确保 GPX4 正常发挥功能是一个有趣的悬而未决的问题。
Ferroptosis regulatory activities occurring in the cytosol
细胞质中发生的铁死亡调节活动
Ferroptosis execution centres on the plasma membrane. However, key ferroptosis regulatory activities occur in the cytosol. The translation of selenoproteins such as GPX4 occurs in the cytosol. Selenocysteine is synthesized directly on a dedicated tRNA (tRNA[Ser]Sec) in a multistep process. tRNA[Ser]Sec is first conjugated with serine, which is then phosphorylated, allowing for the subsequent incorporation of selenium from selenophosphate and yielding the selenocysteine-conjugated tRNA. Selenoprotein transcripts are unique for two reasons. First, they contain a UGA codon within the open reading frame. UGA codons are typically stop codons that bind release factors to terminate translation. However, in selenoprotein transcripts, these UGA codons are recoded for selenocysteine insertion. Second, they contain a stem-loop-like structure called the selenocysteine insertion sequence (SECIS) element in the 3′ untranslated region that recruits machinery such as SECIS binding protein 2 to recode the ribosome for insertion of selenocysteine at the UGA codon, rather than the binding of a ribosome release factor. This unique mode of translation makes selenoprotein translation less efficient and prone to early termination, especially under conditions of limiting selenium63,67.
铁死亡的执行中心是质膜。然而,关键的铁死亡调节活动发生在细胞质中。硒蛋白(例如 GPX4)的翻译发生在细胞质中。硒代半胱氨酸是通过多步骤过程直接在专用 tRNA (tRNA [Ser]Sec ) 上合成的。 tRNA [Ser]Sec首先与丝氨酸缀合,然后磷酸化,随后从硒磷酸盐中掺入硒并产生硒代半胱氨酸缀合的 tRNA。硒蛋白转录物之所以独特有两个原因。首先,它们在开放阅读框中包含UGA密码子。 UGA密码子通常是结合释放因子以终止翻译的终止密码子。然而,在硒蛋白转录物中,这些 UGA 密码子被重新编码以插入硒代半胱氨酸。其次,它们在 3' 非翻译区包含一个称为硒代半胱氨酸插入序列 (SECIS) 元件的茎环样结构,该元件招募 SECIS 结合蛋白 2 等机制来重新编码核糖体,以便在 UGA 密码子处插入硒代半胱氨酸,而不是核糖体释放因子的结合。这种独特的翻译模式使得硒蛋白翻译效率较低,并且容易提前终止,特别是在限制硒的条件下63 , 67 。
Important anti-ferroptotic metabolites are also synthesized in the cytosol. The GPX4 cofactor GSH is synthesized in a two-step reaction by glutamate cysteine ligase (GCL; comprising a catalytic and a modifier subunit, GCLC and GCLM, respectively) and GSH synthetase (GSS). Cysteine, obtained from imported cystine or other sources, is rate-limiting for GSH synthesis. It is often assumed that GSH alone is sufficient to inhibit ferroptosis. However, this might not be the case. Genetic disruption of GCLC does not seem to induce ferroptosis despite eliminating de novo GSH synthesis68. It seems possible that one or more additional cysteine-derived, sulfur-containing metabolites can suppress ferroptosis, in parallel to GSH. For example, co-depletion of GSH and the metabolic intermediate coenzyme A (CoA) is necessary and sufficient to trigger ferroptosis in some cancer cells69,70. How CoA impacts ferroptosis is unclear but could be related to the requirement for CoA in lipid metabolism or to a direct role for CoA in modulating protein function via post-translational modification (that is, CoAlation). Additionally, hydropersulfides can suppress lipid peroxidation71,72, and the synthesis of these metabolites requires cysteine. A further possibility is that GSH tripeptide (γ-glutamylcysteinylglycine) serves as a sink for glutamate, which otherwise can act in a pro-ferroptotic manner. Under conditions of intracellular cysteine starvation, GCL remains active and synthesizes an array of alternative dipeptides and tripeptides that contain glutamate and different amino acids in place of cysteine (for example, γ-glutamyl-threonine)73. The synthesis of these unusual metabolites removes free glutamate from the cell, which otherwise enhances pro-ferroptotic oxidative stress through a downstream mechanism that requires further elicidation73.
重要的抗铁死亡代谢物也在细胞质中合成。 GPX4 辅因子 GSH 通过谷氨酸半胱氨酸连接酶(GCL;分别包含催化亚基和修饰亚基,GCLC 和 GCLM)和 GSH 合成酶 (GSS) 进行两步反应合成。从进口胱氨酸或其他来源获得的半胱氨酸是谷胱甘肽合成的限速剂。人们通常认为单独使用 GSH 就足以抑制铁死亡。然而,情况可能并非如此。尽管消除了谷胱甘肽从头合成68 ,但 GCLC的遗传破坏似乎不会诱导铁死亡。一种或多种额外的半胱氨酸衍生的含硫代谢物似乎可能与 GSH 一样抑制铁死亡。例如,GSH 和代谢中间体辅酶 A (CoA) 的共同消耗对于触发某些癌细胞中的铁死亡是必要且充分的69、70 。 CoA 如何影响铁死亡尚不清楚,但可能与脂质代谢中 CoA 的需求或 CoA 通过翻译后修饰(即 CoAlation)调节蛋白质功能的直接作用有关。此外,氢过硫化物可以抑制脂质过氧化71、72 ,并且这些代谢物的合成需要半胱氨酸。另一种可能性是 GSH 三肽(γ-谷氨酰半胱氨酰甘氨酸)充当谷氨酸的接收器,否则谷氨酸可以以促铁死亡的方式发挥作用。在细胞内半胱氨酸饥饿的条件下,GCL 保持活性并合成一系列替代二肽和三肽,其中含有谷氨酸和不同氨基酸代替半胱氨酸(例如,γ-谷氨酰-苏氨酸) 73 。 这些不寻常代谢物的合成会从细胞中去除游离谷氨酸,否则会通过需要进一步阐明的下游机制增强促铁死亡氧化应激73 。
NADPH is a key electron carrier in the cell. NADP can be synthesized in the cytosol from nicotinamide adenine dinucleotide (NAD+) by NAD kinase (NADK) and then reduced to NADPH by malic enzyme 1 (ME1), isocitrate dehydrogenase (IDH1), or enzymes in the oxidative pentose phosphate pathway74. Higher NADPH levels generally correlate with greater ferroptosis resistance75; this is sensible as NADPH is used to synthesize or regenerate endogenous antioxidant metabolites such as GSH from oxidized glutathione (GSSG) by glutathione-disulfide reductase (GSR), reduced CoQ from oxidized CoQ and reduced vitamin K from oxidized vitamin K by FSP1 (refs. 41,42), and reduced tetrahydrobiopterin (BH4) from BH2 by dihydrofolate reductase (DHFR)23,45. Notably, however, NADPH is also an electron donor for the synthesis of ROS by NOX enzymes at the plasma membrane and by the oxidoreductases cytochrome P450 oxidoreductase (POR) and cytochrome b5 reductase 1 (CYB5R1) at the ER membrane76,77. Thus, NADPH likely has both pro-ferroptotic and anti-ferroptotic functions whose relative importance will vary between cells.
NADPH 是细胞中关键的电子载体。 NADP 可以在细胞质中通过 NAD 激酶 (NADK) 从烟酰胺腺嘌呤二核苷酸 (NAD + ) 合成,然后通过苹果酸酶 1 (ME1)、异柠檬酸脱氢酶 (IDH1) 或氧化戊糖磷酸途径中的酶还原为 NADPH 74 。较高的 NADPH 水平通常与较高的铁死亡抗性相关75 ;这是合理的,因为 NADPH 用于合成或再生内源性抗氧化代谢物,例如通过谷胱甘肽二硫化物还原酶 (GSR) 从氧化型谷胱甘肽 (GSSG) 中合成 GSH,通过 FSP1 从氧化 CoQ 中还原 CoQ,以及从氧化维生素 K 中还原维生素 K(参考文献 1)。 41 , 42 ),以及从 BH2 还原的四氢生物蝶呤 (BH4)通过二氢叶酸还原酶 (DHFR) 23 , 45 。然而,值得注意的是,NADPH还是质膜上的NOX酶以及ER膜上的氧化还原酶细胞色素P450氧化还原酶(POR)和细胞色素b5还原酶1(CYB5R1)合成ROS的电子供体76、77 。因此,NADPH 可能同时具有促铁死亡和抗铁死亡的功能,其相对重要性因细胞而异。
Recent studies connect NADPH to ferroptosis in unexpected ways. HD domain-containing 3 (HDDC3; also known as MESH1) is proposed to function as an NADPH phosphatase whose activity enhances ferroptosis sensitivity by decreasing the levels of NADPH78. Experiments manipulating cytosolic NADK and mitochondrial NADK2 establish that cytosolic NADPH is most critical for governing ferroptosis sensitivity78. NADPH may also be directly sensed by an ER-localized enzyme. The ER-localized E3 ubiquitin ligase membrane-associated ring-CH-type finger 6 (MARCHF6) may directly sense cytosolic NADPH levels via a unique C-terminal binding region, increasing the activity of this enzyme and resulting in changes in the levels of key regulators of ferroptosis, including acyl-CoA synthetase long-chain family member 4 (ACSL4) and p53 (ref. 79). Remarkably, embryonic development is impaired in Marchf6–/– animals and this can be partially reverted by a maternal diet enriched in the natural RTA vitamin E. Thus, MARCHF6 may play a role in preventing ferroptosis during development.
最近的研究以意想不到的方式将 NADPH 与铁死亡联系起来。含 HD 结构域 3(HDDC3;也称为 MESH1)被认为具有 NADPH 磷酸酶的功能,其活性通过降低 NADPH 水平来增强铁死亡敏感性78 。操纵细胞质 NADK 和线粒体 NADK2 的实验表明,细胞质 NADPH 对于控制铁死亡敏感性最为关键78 。 NADPH 也可以被内质网定位的酶直接感知。 ER 定位的 E3 泛素连接酶膜相关环 CH 型指 6 (MARCHF6) 可以通过独特的 C 端结合区域直接感测胞质 NADPH 水平,从而增加该酶的活性并导致关键酶水平的变化。铁死亡的调节因子,包括酰基辅酶A合成酶长链家族成员4(ACSL4)和p53(参考文献79 )。值得注意的是, Marchf6 –/–动物的胚胎发育受到损害,而这种情况可以通过富含天然 RTA 维生素 E 的母体饮食部分恢复。因此,MARCHF6 可能在发育过程中预防铁死亡中发挥作用。
The role of specific organelles in modulating ferroptosis
特定细胞器在调节铁死亡中的作用
Most organelles have been linked to the regulation of ferroptosis sensitivity in one way or another, and often in several potentially contradictory ways at once. How these different and sometimes opposing activities are coordinated to generate a unified effect on ferroptosis in a given context is an important open question. Key aspects of ferroptosis regulation by different organelles are described below.
大多数细胞器都以一种或另一种方式与铁死亡敏感性的调节相关,并且常常同时以几种潜在矛盾的方式相关。如何协调这些不同的、有时是相反的活动,以在给定的背景下对铁死亡产生统一的影响是一个重要的悬而未决的问题。不同细胞器铁死亡调节的关键方面如下所述。
Mitochondria 线粒体
Mitochondria are multifunctional organelles that can regulate ferroptosis sensitivity in several different ways (Fig. 2). In cells treated with the system xc– inhibitor erastin, mitochondria are smaller, with disorganized cristae1,80. This represents a potential morphological marker of ferroptosis. However, it is not clear whether changes in mitochondrial morphology contribute to the execution of ferroptosis or are merely a correlate of this process. For example, mitochondria can be eliminated from some cells with little effect on ferroptosis sensitivity81. In other cells, mitochondria seem to be essential for ferroptosis82. Below, we describe examples of the pro-ferroptotic and anti-ferroptotic effects of mitochondria, touching on specific examples of context-dependent effects and areas of disagreement in the literature.
线粒体是多功能细胞器,可以通过几种不同的方式调节铁死亡敏感性(图2 )。在用系统 x c –抑制剂erastin 处理的细胞中,线粒体较小,嵴混乱1 , 80 。这代表了铁死亡的潜在形态学标记。然而,尚不清楚线粒体形态的变化是否有助于铁死亡的执行,或者仅仅是该过程的相关因素。例如,可以从一些细胞中消除线粒体,而对铁死亡敏感性影响很小81 。在其他细胞中,线粒体似乎对于铁死亡至关重要82 。下面,我们描述线粒体的促铁死亡和抗铁死亡作用的例子,涉及背景依赖性效应的具体例子和文献中存在分歧的领域。
图 2:线粒体中的铁死亡调节。

Mitochondria play several context-dependent roles in the regulation of ferroptosis sensitivity. a, Metabolic reactions — the breakdown of sugar and amino acids via glycolysis and the tricarboxylic acid (TCA) cycle yields NADH and FADH2, which pass electrons to the mitochondrial electron transport chain (ETC). The ETC can partially reduce oxygen to generate reactive oxygen species (ROS) such as superoxide (O2•−) at complexes I and III. Dismutation of O2•− yields hydrogen peroxide, which can result in the formation of hydroxyl radicals (HO•) via reactions with Fe2+ in the Fenton reaction. Glutamine can be catabolized to glutamate (a substrate for the system xc– antiporter) and then to the TCA cycle intermediate α-ketoglutarate (αKG). αKG synthesis promotes ferroptosis, possibly by enhancing mitochondrial ROS production. b, Iron handling — mitoferrin 1 (MFRN1) and MFRN2 mediate iron transport across the inner mitochondrial membrane. The synthesis of Fe–S clusters and haem prosthetic groups in the mitochondria consumes labile iron that otherwise may accumulate to promote ROS accumulation. c, Mitochondrial integrated stress response — OMA1-dependent proteolytic processing of DAP3 binding cell death enhancer 1 (DELE1) releases a protein fragment that initiates the integrated stress response, culminating in activating transcription factor 4 (ATF4)-dependent transcription of a programme that enhances glutathione metabolism and protects against ferroptosis. d, Coenzyme Q10 (CoQ) synthesis — the final steps in de novo CoQ synthesis occur in the mitochondria. CoQ is employed as a key electron carrier in the ETC. CoQ can be reduced by dihydroorotate dehydrogenase (DHODH) to CoQH2, which can either transfer electrons to complex III in the ETC or function as a local radical-trapping antioxidant to prevent peroxidation of mitochondrial lipids and limit ferroptosis. In addition, CoQ is trafficked by a processed form of StAR-related lipid transfer protein 7 (STARD7) from the mitochondria to the plasma membrane, where it can contribute to ferroptosis protection mediated by ferroptosis suppressor protein 1. CoA, coenzyme A.
线粒体在铁死亡敏感性的调节中发挥着多种背景依赖性作用。 a ,代谢反应 — 糖和氨基酸通过糖酵解和三羧酸 (TCA) 循环分解,产生 NADH 和 FADH 2 ,将电子传递到线粒体电子传递链 (ETC)。 ETC可以部分还原氧以在配合物I和III处产生活性氧(ROS),例如超氧化物(O 2 •− )。 O 2 •−的歧化产生过氧化氢,在芬顿反应中与 Fe 2+反应可导致羟基自由基 (HO•) 的形成。谷氨酰胺可分解代谢为谷氨酸(x c系统的底物-逆向转运蛋白),然后分解为 TCA 循环中间体 α-酮戊二酸 (αKG)。 αKG 合成可能通过增强线粒体 ROS 产生来促进铁死亡。 b ,铁处理——线粒体铁蛋白 1 (MFRN1) 和 MFRN2 介导铁穿过线粒体内膜的转运。线粒体中 Fe-S 簇和血红素辅基的合成会消耗不稳定的铁,否则这些铁可能会积累以促进 ROS 积累。 c ,线粒体整合应激反应 — DAP3 结合细胞死亡增强子 1 (DELE1) 的 OMA1 依赖性蛋白水解过程释放启动整合应激反应的蛋白片段,最终激活转录因子 4 (ATF4) 依赖的程序转录,从而增强细胞死亡谷胱甘肽代谢并防止铁死亡。 d ,辅酶 Q10 (CoQ) 合成 — CoQ 从头合成的最后步骤发生在线粒体中。 CoQ 被用作 ETC 中的关键电子载体。 CoQ 可以被二氢乳清酸脱氢酶 (DHODH) 还原为 CoQH 2 ,CoQH 2 可以将电子转移到 ETC 中的复合物 III,或者充当局部自由基捕获抗氧化剂,以防止线粒体脂质过氧化并限制铁死亡。此外,CoQ 通过加工形式的 StAR 相关脂质转移蛋白 7 (STARD7) 从线粒体运输到质膜,在质膜中它可以促进铁死亡抑制蛋白 1 介导的铁死亡保护。CoA,辅酶 A。
Several mitochondrial activities reduce ferroptosis sensitivity. Mitofusin 1 (MFN1)-mediated mitochondrial fusion may reduce sensitivity to RSL3-induced ferroptosis, at least in cultured cancer cells83. Likewise, in cells with dysfunctional mitochondria, activation of the mitochondrial stress response pathway mediated by metalloendopeptidase OMA1 and DAP3 binding cell death enhancer 1 (DELE1) can inhibit ferroptosis and delay cardiomyopathy84. Here, DELE1 activates cytosolic activating transcription factor 4 (ATF4), which in turn promotes GSH synthesis and GPX4 protein stability. Mitochondria are also the site of CoQ metabolite synthesis85. CoQ is an electron carrier in the mitochondrial electron transport chain (ETC) but also functions as an important anti-ferroptotic RTA41,42. Key enzymatic machinery necessary for CoQ synthesis (for example, the COQ proteins) localize to the mitochondria. A specific proteolytically processed version of StAR-related lipid transfer protein 7 (STARD7) transports CoQ from the mitochondria to the plasma membrane, where it can be reduced by FSP1 to inhibit lipid peroxidation and ferroptosis86. Whether STARD7 might act at membrane contact sites between the mitochondria and plasma membrane to promote efficient and directional CoQ transfer is unknown. Another important protective role for mitochondria is in the breakdown of PUFAs that could otherwise be incorporated into membrane phospholipids, peroxidized and thereby promote ferroptosis. This catabolic process requires the mitochondrial enzyme 2,4-dienoyl-CoA reductase 1 (DECR1) and, for reasons that remain unclear, appears especially relevant in prostate cancer87,88. Collectively, these mitochondrial functions would tend to reduce ferroptosis sensitivity.
几种线粒体活性可降低铁死亡敏感性。线粒体融合蛋白 1 (MFN1) 介导的线粒体融合可能会降低对 RSL3 诱导的铁死亡的敏感性,至少在培养的癌细胞中是这样83 。同样,在线粒体功能障碍的细胞中,金属内肽酶 OMA1 和 DAP3 结合细胞死亡增强子 1 (DELE1) 介导的线粒体应激反应途径的激活可以抑制铁死亡并延缓心肌病84 。在这里,DELE1 激活胞质激活转录因子 4 (ATF4),进而促进 GSH 合成和 GPX4 蛋白稳定性。线粒体也是 CoQ 代谢物合成的场所85 。 CoQ 是线粒体电子传递链 (ETC) 中的电子载体,同时也是重要的抗铁死亡 RTA 41 、 42 。 CoQ 合成所需的关键酶机制(例如 COQ 蛋白)位于线粒体。 StAR 相关脂质转移蛋白 7 (STARD7) 的特定蛋白水解加工版本将 CoQ 从线粒体转运到质膜,在质膜中它可以被 FSP1 还原,从而抑制脂质过氧化和铁死亡86 。 STARD7 是否可能作用于线粒体和质膜之间的膜接触位点以促进有效和定向的 CoQ 转移尚不清楚。线粒体的另一个重要保护作用是分解多不饱和脂肪酸,否则多不饱和脂肪酸可能会掺入膜磷脂中,发生过氧化,从而促进铁死亡。 这种分解代谢过程需要线粒体酶 2,4-二烯酰辅酶 A 还原酶 1 (DECR1),并且由于尚不清楚的原因,它似乎与前列腺癌特别相关87 , 88 。总的来说,这些线粒体功能往往会降低铁死亡的敏感性。
Other mitochondrial mechanisms increase ferroptosis sensitivity. For example, the mitochondrial tricarboxylic acid (TCA) cycle promotes ferroptosis sensitivity, at least in response to cystine deprivation89. During glutaminolysis, glutamine is converted to glutamate and the TCA cycle intermediate α-ketoglutarate within mitochondria; both glutamate and α-ketoglutarate can promote ferroptosis53,73,90. In these cases, the precise effector mechanism linking TCA cycle activity with the induction of ferroptosis remains obscure, although enhanced oxidative stress seems to be involved. Mitochondria are also central hubs for the use and metabolism of iron2. Increased mitochondrial iron uptake mediated by mitoferrin 1 (MFRN1; encoded by SLC25A37) can enhance ferroptosis sensitivity, although the exact mechanism is unclear91. Disruption of mitochondrial biosynthesis of Fe–S clusters can also enhance ferroptosis sensitivity. Here, depletion of NFS1 cysteine desulfurase lowers the synthesis of Fe–S clusters, triggering a feedback iron starvation response that increases iron import and heightens ferroptosis sensitivity92. It might be the case that cystine deprivation caused by system xc– inhibition has unique effects on mitochondrial Fe–S biosynthesis given the need for cysteine-derived sulfur in this process. One model to explain observed differences in the ferroptosis phenotypes caused by cystine deprivation versus direct GPX4 inhibition10 is that cystine deprivation uniquely perturbs mitochondrial Fe–S cluster biosynthesis45. Finally, mitochondrial outer membrane permeabilization, when it occurs in a partial and non-lethal manner throughout the cell, results in both activation of the ATF4 pathway and enhanced sensitivity to ferroptosis induced by GPX4 inhibitors93. This may suggest that the loss of one or more mitochondrial anti-ferroptotic functions can, on balance, overcome the protective effect of ATF4 pathway activation.
其他线粒体机制增加了铁死亡的敏感性。例如,线粒体三羧酸 (TCA) 循环会促进铁死亡敏感性,至少是对胱氨酸剥夺的反应89 。在谷氨酰胺分解过程中,谷氨酰胺在线粒体内转化为谷氨酸和 TCA 循环中间体 α-酮戊二酸;谷氨酸和α-酮戊二酸均可促进铁死亡53 , 73 , 90 。在这些情况下,尽管似乎涉及增强的氧化应激,但将 TCA 循环活性与铁死亡诱导联系起来的精确效应机制仍然不清楚。线粒体也是铁的利用和代谢的中心枢纽2 。线粒体铁蛋白 1(MFRN1;由SLC25A37编码)介导的线粒体铁摄取增加可以增强铁死亡敏感性,但确切机制尚不清楚91 。线粒体Fe-S 簇生物合成的破坏也可以增强铁死亡的敏感性。在这里,NFS1 半胱氨酸脱硫酶的消耗降低了 Fe-S 簇的合成,触发反馈铁饥饿反应,增加铁输入并提高铁死亡敏感性92 。考虑到在此过程中需要半胱氨酸衍生的硫,系统 x c抑制引起的胱氨酸剥夺可能对线粒体 Fe-S 生物合成具有独特的影响。 一种解释胱氨酸剥夺与直接 GPX4 抑制引起的铁死亡表型差异的模型10是胱氨酸剥夺独特地扰乱线粒体 Fe-S 簇生物合成45 。最后,当线粒体外膜透化在整个细胞中以部分和非致命的方式发生时,会导致 ATF4 途径的激活并增强对 GPX4 抑制剂诱导的铁死亡的敏感性93 。这可能表明,一种或多种线粒体抗铁死亡功能的丧失总体上可以克服 ATF4 途径激活的保护作用。
In some cancer cells, mitochondrial ETC activity promotes ferroptosis, at least in response to cystine deprivation, as shown using specific ETC poisons89. How the ETC promotes ferroptosis is not clear. One obvious candidate is the production of ROS that might be expected to initiate lipid peroxidation. Indeed, ROS scavengers that target mitochondria can attenuate ferroptosis in some models, implying that mitochondria-derived oxidants may contribute to cell death (Box 2). However, in other cancer cells, depleting the mitochondrial DNA, resulting in complete loss of ETC function, has no effect on ferroptosis sensitivity1,94. Moreover, ETC function is necessary for the activity of two mitochondrially localized enzymes that may be able to generate reduced CoQ: dihydroorotate dehydrogenase (DHODH)37 and glycerol-3-phosphate dehydrogenase 2 (GPD2)95. Accordingly, loss of ETC function might be expected to enhance ferroptosis sensitivity by disrupting the synthesis of reduced mitochondrial CoQ. Overall, it seems likely that the role of the mitochondrial ETC varies by cell type and by the nature of the lethal stimulus. An emerging (and controversial)38 concept is that the mitochondria might assume a greater importance in overall ferroptosis regulation when cytosolic anti-ferroptotic systems are disrupted37,39,96. This model of a conditional role for mitochondria depending on circumstances could provide a reconciliation for some conflicting observations.
在一些癌细胞中,线粒体 ETC 活性会促进铁死亡,至少是对胱氨酸剥夺的反应,如使用特定 ETC 毒物所示89 。 ETC 如何促进铁死亡尚不清楚。一个明显的候选者是ROS的产生,预计它可能会引发脂质过氧化。事实上,在某些模型中,针对线粒体的 ROS 清除剂可以减弱铁死亡,这意味着线粒体来源的氧化剂可能会导致细胞死亡(框2 )。然而,在其他癌细胞中,耗尽线粒体 DNA 导致 ETC 功能完全丧失,对铁死亡敏感性没有影响1 , 94 。此外,ETC 功能对于两种可能产生还原 CoQ 的线粒体定位酶的活性是必需的:二氢乳清酸脱氢酶 (DHODH) 37和甘油-3-磷酸脱氢酶 2 (GPD2) 95 。因此,ETC 功能的丧失可能会通过破坏还原线粒体 CoQ 的合成来增强铁死亡敏感性。总体而言,线粒体 ETC 的作用可能因细胞类型和致死刺激的性质而异。一个新兴的(且有争议的) 38概念是,当胞质抗铁死亡系统被破坏时,线粒体可能在整体铁死亡调节中发挥更大的重要性37 , 39 , 96 。这种线粒体根据环境发挥条件作用的模型可以为一些相互矛盾的观察结果提供调和。
Endoplasmic reticulum 内质网
The ER plays an important role in ferroptosis regulation. POR and CYB5R1 are enzymes that reside in the ER membrane and use NADPH to produce ROS that can promote membrane lipid peroxidation76,77 (Fig. 3a). Indeed, there is good evidence that the ER is one site where membrane lipid peroxidation is initiated during ferroptosis (Box 2). As noted above, whether lipid peroxidation spreads from the ER to the plasma membrane to execute ferroptosis, for example, through vesicular trafficking or phospholipid transfer at membrane contact sites, is an open question.
ER 在铁死亡调节中发挥重要作用。 POR 和 CYB5R1 是驻留在 ER 膜上的酶,它们利用 NADPH 产生 ROS,从而促进膜脂过氧化76 、 77 (图3a )。事实上,有充分的证据表明内质网是铁死亡过程中启动膜脂过氧化的位点之一(框2 )。如上所述,脂质过氧化是否从内质网扩散到质膜以执行铁死亡,例如通过膜接触位点的囊泡运输或磷脂转移,是一个悬而未决的问题。
图 3:ER 中的铁死亡调节。

a, Cytochrome P450 oxidoreductase (POR) and cytochrome b5 reductase 1 (CYB5R1) are endoplasmic reticulum (ER) enzymes that promote ferroptosis by generating H2O2, which can further react with iron to produce reactive oxygen species species that initiate lipid peroxidation, leading to ferroptosis. Lipid peroxidation in the ER is an early event in ferroptosis. Whether damage propagates from the ER or accumulates at a slower rate in other membranes is an open question (see also Box 2). b, The ER is the primary site of lipid synthesis, including the synthesis of glycerolipids, namely phospholipids (PLs) and triacylglycerols (TAGs). The incorporation of monounsaturated fatty acids (MUFAs) and polyunsaturated fatty acids (PUFAs) into PLs or TAGs helps establish the overall sensitivity of the cell to lipid peroxidation and ferroptosis. TAGs are stored within lipid droplets that emerge from the outer leaflet of the ER. PLs are trafficked to the plasma membrane via the secretory pathway or transferred at ER–plasma membrane contact sites. Phosphatidylethanolamine (PE) is often oxidized during ferroptosis. The synthesis of PE involves the transfer of phosphatidylserine from the ER to mitochondria, where it is enzymatically converted to PE prior to transport back to the ER. The relative contributions of these PL trafficking pathways to ferroptosis remain to be defined. c, A complex series of enzymatic steps in the ER mediate the synthesis of cholesterol via the mevalonate pathway. Several intermediates in this pathway protect against ferroptosis. Farnesyl pyrophosphate (FPP) is needed to synthesize the endogenous radical-trapping antioxidants coenzyme Q10 (CoQ) and vitamin K in the mitochondria and Golgi, respectively. Isopentenyl pyrophosphate (IPP) is attached to the selenocysteine tRNA as an isopentenyl moiety, a key modification for its function, promoting the synthesis of selenoproteins, including the anti-ferroptotic enzyme glutathione peroxidase 4 (GPX4). 7-Dehydrocholesterol (7DHC) is highly prone to oxidation and can suppress ferroptosis by competing with PL for oxidation. Squalene also suppresses ferroptosis, but the mechanism is not understood. d, Proteolytic processing of the membrane-tethered transcription factors sterol regulatory element-binding protein 1 (SREBP1) and nuclear factor erythroid 2-related factor 1 (NFE2L1) releases soluble, active transcription factors that traffic to the nucleus and initiate transcriptional programmes to control lipid metabolism and the cellular oxidative stress response. The phosphoinositide 3-kinase (PI3K)–mechanistic target of rapamycin complex 1 (mTORC1) pathway promotes ferroptosis resistance by increasing SREBP1 activation and expression of stearoyl-CoA desaturase 1 (SCD1), which generates ferroptosis-suppressive MUFAs. NFE2L1 is dislocated from the ER into the cytoplasm, where it is deglycosylated by N-glycanase 1 (NGLY1) and proteolytically processed by DNA-damage inducible 1 homologue 2 (DDI2). The amount of NFE2L1 that escapes proteasomal clearance is a determinant of NFE2L1 transcriptional signalling. HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; MVA, mevalonate; GGPP, geranylgeranyl pyrophosphate; UBIAD1, UBIA prenyltransferase domain-containing protein 1.
a , 细胞色素 P450 氧化还原酶 (POR) 和细胞色素 b5 还原酶 1 (CYB5R1) 是内质网 (ER) 酶,通过产生 H 2 O 2促进铁死亡,H 2 O 2 可以进一步与铁反应产生活性氧物种,引发脂质过氧化,导致铁死亡。内质网中的脂质过氧化是铁死亡的早期事件。损伤是从内质网传播还是以较慢的速度在其他膜中累积是一个悬而未决的问题(另见框2 )。 b ,内质网是脂质合成的主要场所,包括甘油脂的合成,即磷脂(PL)和三酰甘油(TAG)。将单不饱和脂肪酸 (MUFA) 和多不饱和脂肪酸 (PUFA) 纳入 PL 或 TAG 有助于建立细胞对脂质过氧化和铁死亡的整体敏感性。 TAG 储存在从 ER 外层出现的脂滴中。 PL 通过分泌途径运输至质膜或在内质网-质膜接触位点转移。磷脂酰乙醇胺 (PE) 在铁死亡过程中经常被氧化。 PE 的合成涉及将磷脂酰丝氨酸从 ER 转移到线粒体,在线粒体中酶促转化为 PE,然后运回 ER。这些 PL 运输途径对铁死亡的相对贡献仍有待确定。 c ,内质网中一系列复杂的酶促步骤通过甲羟戊酸途径介导胆固醇的合成。该途径中的几种中间体可以防止铁死亡。 法尼基焦磷酸 (FPP) 是线粒体和高尔基体中分别合成内源性自由基捕获抗氧化剂辅酶 Q10 (CoQ) 和维生素 K 所必需的。异戊烯焦磷酸 (IPP) 作为异戊烯基部分附着在硒代半胱氨酸 tRNA 上,这是其功能的关键修饰,可促进硒蛋白的合成,包括抗铁死亡酶谷胱甘肽过氧化物酶 4 (GPX4)。 7-脱氢胆固醇 (7DHC) 极易氧化,可通过与 PL 竞争氧化来抑制铁死亡。角鲨烯也能抑制铁死亡,但其机制尚不清楚。 d ,膜束缚转录因子甾醇调节元件结合蛋白 1 (SREBP1) 和核因子红细胞 2 相关因子 1 (NFE2L1) 的蛋白水解过程释放可溶性活性转录因子,这些转录因子运输到细胞核并启动转录程序来控制脂质代谢和细胞氧化应激反应。磷酸肌醇 3 激酶 (PI3K)——雷帕霉素复合物 1 (mTORC1) 通路的机制靶点通过增加 SREBP1 的激活和硬脂酰辅酶 A 去饱和酶 1 (SCD1) 的表达来促进铁死亡抵抗,从而产生铁死亡抑制性 MUFA。 NFE2L1 从 ER 移位到细胞质中,在细胞质中被 N-聚糖酶 1 (NGLY1) 去糖基化,并被 DNA 损伤诱导型 1 同源物 2 (DDI2) 进行蛋白水解加工。逃避蛋白酶体清除的 NFE2L1 量是 NFE2L1 转录信号传导的决定因素。 HMG-CoA,3-羟基-3-甲基戊二酰辅酶A; MVA,甲羟戊酸; GGPP,香叶基香叶基焦磷酸; UBIAD1,UBIA 异戊烯基转移酶结构域含蛋白 1。
Ferroptosis is a lipid-dependent process. The ER is a major hub for lipid metabolism in the cell. Lipid desaturation, phospholipid synthesis, phospholipid remodelling and biogenesis of lipid droplets are all ER-localized activities (Fig. 3b). Thus, it is no surprise that ER-resident lipid metabolic enzymes contribute importantly to the regulation of ferroptosis sensitivity. Overall, the ferroptotic threshold of the cell can be determined by lipid composition and, more specifically, by the relative levels of different lipid species97. Decreasing the ratio of more oxidizable PUFAs to less oxidizable monounsaturated fatty acids (MUFAs) is sufficient to convert cells from ferroptosis sensitive to ferroptosis resistant28,98,99,100,101 whereas increasing the PUFA to MUFA ratio can have the opposite effect22,77. ER-resident enzymes shape the PUFA and MUFA content of cellular lipid pools. For example, fatty acid desaturase 1 (FADS1) and FADS2, which introduce carbon–carbon double bonds into fatty acids, can enhance or reduce ferroptosis sensitivity, respectively, by synthesizing specific PUFAs or MUFAs102,103. The overall activity of these desaturation processes can be governed within the ER by calcium levels, which are regulated by the tetraspanin membrane-spanning 4-domains subfamily A member 15 (MS4A15)104.
铁死亡是一个脂质依赖性过程。 ER 是细胞内脂质代谢的主要枢纽。脂质去饱和、磷脂合成、磷脂重塑和脂滴的生物发生都是内质网局部的活动(图3b )。因此,内质网驻留的脂质代谢酶对铁死亡敏感性的调节发挥重要作用也就不足为奇了。总体而言,细胞的铁死亡阈值可以通过脂质组成来确定,更具体地说,可以通过不同脂质种类的相对水平来确定97 。降低更多可氧化的 PUFA 与更少氧化的单不饱和脂肪酸(MUFA) 的比例足以将细胞从铁死亡敏感转化为铁死亡抗性28 , 98 , 99 , 100 , 101而增加 PUFA 与 MUFA 的比例可能会产生相反的效果22 , 77 .内质网驻留酶决定细胞脂质库中 PUFA 和 MUFA 的含量。例如,将碳碳双键引入脂肪酸的脂肪酸去饱和酶1 (FADS1)和FADS2可以通过合成特定的PUFA或MUFA 102 、 103分别增强或降低铁死亡敏感性。这些去饱和过程的总体活性可以在 ER 内由钙水平控制,钙水平由四跨膜蛋白 4 结构域亚家族 A 成员 15 (MS4A15) 104调节。
Acyl-CoA synthetase long-chain (ACSL) enzymes ‘activate’ both PUFAs and MUFAs to PUFA-CoA and MUFA-CoA species, which can then be incorporated into membrane phospholipids, triacylglycerols (TAGs) or other lipids. ACSL4 and ACSL3 appear generally to function in opposition, with ACSL4-driven PUFA metabolism increasing ferroptosis sensitivity and ACSL3-dependent MUFA metabolism promoting ferroptosis resistance24,28,30,99,105,106. However, exceptions to this general scheme exist, for example, the pro-ferroptotic role of ACSL3 in KRAS-mutant lung cancer cells107. Further work is required to establish where in the cell these enzymes function to regulate ferroptosis. Genetic rescue data suggests that ER-localized ACSL4 may be critical for ferroptosis regulation27. However, ACSL4 can also sometimes be found in lipid droplets, the Golgi apparatus or at the plasma membrane24,108,109. ACSL3 can also localize to both the ER and to lipid droplets110. Which of these localizations is most important for ferroptosis regulation remains somewhat mysterious and could differ by cell type. How the activities of these enzymes at the ER impact plasma membrane lipid composition and peroxidation27 is also not at all clear.
酰基辅酶 A 合成酶长链 (ACSL) 酶可将 PUFA 和 MUFA“激活”为 PUFA-CoA 和 MUFA-CoA 物质,然后将其掺入膜磷脂、三酰甘油 (TAG) 或其他脂质中。 ACSL4 和 ACSL3 通常表现出相反的功能,ACSL4 驱动的 PUFA 代谢增加铁死亡敏感性,ACSL3 依赖性 MUFA 代谢促进铁死亡抵抗24 , 28 , 30 , 99 , 105 , 106 。然而,这个一般方案存在例外,例如,ACSL3 在KRAS突变肺癌细胞中的促铁死亡作用107 。需要进一步的工作来确定这些酶在细胞中的何处发挥调节铁死亡的作用。基因拯救数据表明,ER 定位的 ACSL4 可能对于铁死亡调节至关重要27 。然而,ACSL4有时也可以在脂滴、高尔基体或质膜24、108、109处发现。 ACSL3还可以定位于ER和脂滴110 。这些定位中哪一个对于铁死亡调节最重要仍然有些神秘,并且可能因细胞类型而异。这些内质网酶的活性如何影响质膜脂质组成和过氧化作用27也完全不清楚。
The ER is the site of de novo phospholipid synthesis111. There is evidence from large-scale genetic screens that ER-resident enzymes involved in de novo phospholipid synthesis, including glycerol-3-phosphate acyltransferase 4 (GPAT4), can promote ferroptosis sensitivity27,111. However, the role of de novo phospholipid synthesis is less explored than the role of phospholipid remodelling (that is, the Lands cycle). The Lands cycle involves cleavage of one acyl chain of a phospholipid by a phospholipase, followed by reacylation of the lysophospholipid with an acyl-CoA to reform the phospholipid. Calcium-independent phospholipase A2 (also known as PLA2G6, PNPLA9 or iPLA2β) can cleave oxidized PUFA acyl chains from phospholipids, thereby directly limiting the spread of lipid peroxidation and the onset of ferroptosis112,113,114. A related enzyme, PLA2G4C, may also negatively regulate ferroptosis in KRAS-mutant lung cancer in a similar manner107. A distinct lysophospholipase enzyme, abhydrolase domain-containing protein 12 (ABHD12), cleaves PUFA-containing lysophosphatidylserine and oxidized phosphatidylserines specifically, and inhibition of this enzyme is sufficient to increase PUFA-phosphatidylserine (that is, 18:0/20:4 phosphatidylserine) levels and boost ferroptosis sensitivity. Lysophospholipids are reacylated (or, in de novo synthesis, acylated) by phospholipid acyltransferases22. For example, reacylation of lysophospholipids with PUFAs is catalysed by ER-resident enzymes, including lysophosphatidylcholine acyltransferase 3 (LPCAT3; also known as MBOAT5), 1-acylglycerol-3-phosphate O-acyltransferase 3 (AGPAT3)25,105,115 and MBOAT7 (ref. 24), while ER-resident MBOAT1 and MBOAT2 acylate lysophospholipids with MUFAs116,117. Presumably, there is competition between phospholipid acyltransferases to reacylate (or acylate) the same substrates, and the relative levels of different enzymes and substrates will help determine the overall composition of the membrane and, in turn, the ferroptosis sensitivity of the cell. Although LPCAT3 is an ER-resident enzyme118, the phospholipase PLA2G6 may be found through the cytosol with the possibility of being recruited to the vicinity of the plasma membrane119. Thus, it is also unclear whether all ferroptosis-relevant lipid synthesis and remodelling events occur in the ER, or whether localized remodelling in other endomembrane compartments and/or at the plasma membrane also helps shape ferroptosis sensitivity.
ER 是磷脂从头合成的位点111 。大规模遗传筛选的证据表明,参与磷脂从头合成的内质网驻留酶,包括甘油-3-磷酸酰基转移酶 4 (GPAT4),可以促进铁死亡敏感性27 , 111 。然而,与磷脂重塑(即 Lands 循环)的作用相比,磷脂从头合成的作用较少被研究。 Lands 循环涉及磷脂酶裂解磷脂的一条酰基链,然后用酰基辅酶 A 将溶血磷脂再酰化以重新形成磷脂。钙非依赖性磷脂酶 A2(也称为 PLA2G6、PNPLA9 或 iPLA2β)可以从磷脂中裂解氧化的 PUFA 酰基链,从而直接限制脂质过氧化的扩散和铁死亡的发生112 , 113 , 114 。一种相关酶 PLA2G4C 也可能以类似的方式负向调节KRAS突变型肺癌中的铁死亡107 。一种独特的溶血磷脂酶,含脱水酶结构域的蛋白 12 (ABHD12),可特异性裂解含有 PUFA 的溶血磷脂酰丝氨酸和氧化磷脂酰丝氨酸,抑制该酶足以增加 PUFA-磷脂酰丝氨酸(即 18:0/20:4 磷脂酰丝氨酸)水平并提高铁死亡敏感性。溶血磷脂被磷脂酰基转移酶重新酰化(或在从头合成中酰化) 22 。 例如,溶血磷脂与 PUFA 的再酰化由内质网驻留酶催化,包括溶血磷脂酰胆碱酰基转移酶 3 (LPCAT3;也称为 MBOAT5)、1-酰基甘油-3-磷酸 O-酰基转移酶 3 (AGPAT3) 25 、 105 、 115和 MBOAT7 (参考文献24 ),而内质网驻留的 MBOAT1 和 MBOAT2 用 MUFA 酰化溶血磷脂116 、 117 。据推测,磷脂酰基转移酶之间存在竞争以重新酰化(或酰化)相同的底物,并且不同酶和底物的相对水平将有助于确定膜的整体组成,进而确定细胞的铁死亡敏感性。虽然LPCAT3是内质网驻留酶118 ,但是磷脂酶PLA2G6可以通过胞质溶胶被发现,并且有可能被募集至质膜附近119 。因此,还不清楚是否所有与铁死亡相关的脂质合成和重塑事件都发生在内质网中,或者其他内膜区室和/或质膜上的局部重塑是否也有助于塑造铁死亡敏感性。
In addition to enzymes involved in phospholipid metabolism, the ER houses the enzymes of the mevalonate pathway (Fig. 3c). Although this pathway is best known for the synthesis of cholesterol, several intermediates within this pathway are important in ferroptosis suppression. For example, an intermediate in cholesterol synthesis, 7-dehydrocholesterol, is highly prone to oxidation and can act as a sacrificial endogenous RTA to limit membrane lipid peroxidation120,121. A different metabolite, squalene, has also been implicated in ferroptosis suppression122, but the mechanism remains unclear. Another product of the mevalonate pathway is farnesyl pyrophosphate, a key precursor for the synthesis of vitamin K in the Golgi123 and CoQ in the mitochondria85. Finally, another mevalonate pathway product, isopentenyl pyrophosphate is necessary for isopentylation of the Sec-tRNA and it thereby modulates selenoprotein translation. Whether isopentylation impacts GPX4 levels to an extent that would affect ferroptosis sensitivity remains to be directly tested124.
除了参与磷脂代谢的酶外,ER 还含有甲羟戊酸途径的酶(图3c )。尽管该途径以胆固醇的合成而闻名,但该途径中的几种中间体对于抑制铁死亡很重要。例如,胆固醇合成的中间体 7-脱氢胆固醇非常容易氧化,可以作为牺牲性内源性 RTA 来限制膜脂过氧化120、121 。一种不同的代谢物角鲨烯也与铁死亡抑制有关122 ,但其机制仍不清楚。甲羟戊酸途径的另一个产物是法尼基焦磷酸,它是高尔基体中合成维生素 K 123和线粒体中合成 CoQ 85的关键前体。最后,另一种甲羟戊酸途径产物异戊烯焦磷酸对于 Sec-tRNA 的异戊烯化是必需的,从而调节硒蛋白翻译。异戊基化是否会影响 GPX4 水平,从而影响铁死亡敏感性,仍有待直接测试124 。
Beyond its roles in lipid synthesis, the ER is a site for the processing of important transcription factors that modulate the cellular lipid and redox environments (Fig. 3d). Sterol regulatory element-binding proteins (SREBPs) are ER-resident transcription factors that regulate the expression of many enzymes related to lipid metabolism. Tethered to the ER by two transmembrane domains, SREBPs undergo regulated trafficking to the Golgi, where they are proteolytically processed to release the active transcription factor, which traffics to the nucleus to initiate transcription. A signalling pathway involving phosphatidylinositol 3-kinase (PI3K), AKT and mechanistic target of rapamycin complex 1 (mTORC1), can activate SREBP1 to protect cancer cells from ferroptosis125. SREBP1-dependent expression of stearoyl-CoA desaturase 1 (SCD1) is required for this effect125. SCD1 catalyses the synthesis of long-chain MUFAs (for example, oleate); thus, increasing MUFA production should reduce membrane susceptibility to peroxidation and ferroptosis sensitivity. A second transcription factor that is processed via the ER is nuclear factor erythroid 2-related factor 1 (NFE2L1). NFE2L1 undergoes a complex series of post-translational modifications, including glycosylation in the ER and subsequent deglycosylation in the cytosol by N-glycanase 1 (NGLY1), as well as proteolytic cleavage by DNA-damage inducible 1 homologue 2 (DDI2), to yield an active transcription factor126,127,128,129. The NGLY1–NFE2L1 axis helps inhibit ferroptosis in some cells by promoting GPX4 protein stability indirectly, possibly through regulation of proteasome gene expression or function130,131.
除了在脂质合成中的作用之外,内质网还是处理调节细胞脂质和氧化还原环境的重要转录因子的位点(图3d )。甾醇调节元件结合蛋白(SREBP)是内质网驻留转录因子,调节许多与脂质代谢相关的酶的表达。 SREBP 通过两个跨膜结构域与 ER 相连,受调控地运输至高尔基体,在高尔基体中进行蛋白水解加工以释放活性转录因子,该转录因子运输至细胞核以启动转录。涉及磷脂酰肌醇 3 激酶 (PI3K)、AKT 和雷帕霉素复合物 1 (mTORC1) 机制靶标的信号通路可以激活 SREBP1,以保护癌细胞免于铁死亡125 。此效果需要 SREBP1 依赖性硬脂酰辅酶 A 去饱和酶 1 (SCD1) 的表达125 。 SCD1催化长链MUFA(例如油酸)的合成;因此,增加 MUFA 产量应降低膜对过氧化的敏感性和铁死亡的敏感性。通过 ER 处理的第二种转录因子是核因子红细胞 2 相关因子 1 (NFE2L1)。 NFE2L1 经历一系列复杂的翻译后修饰,包括 ER 中的糖基化和随后 N-聚糖酶 1 (NGLY1) 在胞质溶胶中的去糖基化,以及 DNA 损伤诱导型 1 同源物 2 (DDI2) 的蛋白水解切割,从而产生活性转录因子126 , 127 , 128 , 129 。 NGLY1–NFE2L1 轴可能通过调节蛋白酶体基因表达或功能,间接促进 GPX4 蛋白稳定性,从而有助于抑制某些细胞中的铁死亡130 、 131 。
Finally, the overall function of the ER may be impaired in cells undergoing ferroptosis, at least when induced by cystine deprivation. Cystine deprivation can trigger a global integrated stress response and ER stress response, including changes in gene expression and the viscosity and pH of the ER58,132. Whether these changes contribute directly to the execution of ferroptosis is not known. However, certain changes, such as the transcriptional upregulation of SLC7A11 (which encodes the transport subunit of system xc–)58, might be expected to reduce ferroptosis sensitivity, while changes in viscosity or pH could very well modulate the function of one or more ER-resident enzymes or mechanisms noted above. In sum, the ER has a multifaceted role in ferroptosis regulation.
最后,在经历铁死亡的细胞中,内质网的整体功能可能受到损害,至少在胱氨酸剥夺诱导时是如此。胱氨酸剥夺可以触发整体综合应激反应和内质网应激反应,包括基因表达以及内质网粘度和pH值的变化58 , 132 。这些变化是否直接导致铁死亡的执行尚不清楚。然而,某些变化,例如SLC7A11 (编码系统 x c –的转运亚基) 58的转录上调,可能会降低铁死亡敏感性,而粘度或 pH 值的变化可以很好地调节一种或多种功能上述内质网驻留酶或机制。总之,ER 在铁死亡调节中具有多方面的作用。
Lipid droplets 脂滴
Lipid droplets are dynamic ER-derived lipid storage organelles that have a context-specific role in ferroptosis regulation133 (Fig. 4). Lipid droplets consist of a neutral lipid core, composed mostly of TAG and steryl esters, encircled by a phospholipid monolayer containing integral and peripheral regulatory proteins134. Different fatty acids can be stored in or released from lipid droplet-resident lipids with varying effects on ferroptosis sensitivity. For example, cancer cells grown in acidic conditions that mimic the tumour microenvironment accumulate lipid droplets, and inhibition of lipid droplet synthesis channels PUFAs away from TAGs and into phospholipids, sensitizing cells to ferroptosis135. In mice, oral gavage with PUFA-rich oils together with blockade of lipid droplet biosynthesis increases PUFA-containing phospholipids and is sufficient to trigger ferroptosis in tumour xenografts135. Conceptually similar, in patient-derived glioblastoma cells, if PUFAs are not sequestered in lipid droplets away from membrane phospholipids they can promote ferroptosis sensitivity136. Here, storage of PUFAs in lipid droplets versus incorporation into membrane phospholipids is genotype dependent, with CDKN2A-null glioblastoma cells specifically showing reduced PUFA sequestration in lipid droplets and enhanced ferroptosis induction136. Finally, in cell-cycle-arrested cancer cells, PUFAs can be channelled into lipid droplets, promoting ferroptosis resistance137. Thus, in some cells, the preferential sequestration of PUFAs in lipid droplets, away from membrane phospholipids, limits ferroptosis sensitivity.
脂滴是动态的内质网衍生的脂质储存细胞器,在铁死亡调节中具有特定的作用133 (图4 )。脂滴由中性脂质核心组成,主要由TAG和甾醇酯组成,被含有整体和外周调节蛋白134的磷脂单层包围。不同的脂肪酸可以储存在脂滴驻留的脂质中或从脂滴驻留的脂质中释放,对铁死亡敏感性有不同的影响。例如,在模拟肿瘤微环境的酸性条件下生长的癌细胞会积累脂滴,并且抑制脂滴合成会将PUFA从TAG转移到磷脂中,从而使细胞对铁死亡敏感135 。在小鼠中,口服富含 PUFA 的油并阻断脂滴生物合成会增加含 PUFA 的磷脂,并足以引发肿瘤异种移植物中的铁死亡135 。概念上类似,在患者来源的胶质母细胞瘤细胞中,如果 PUFA 没有被隔离在远离膜磷脂的脂滴中,它们可以促进铁死亡敏感性136 。此处,PUFA 在脂滴中的储存与掺入膜磷脂中的情况是基因型依赖性的, CDKN2A缺失的胶质母细胞瘤细胞特别显示出 PUFA 在脂滴中的隔离减少和铁死亡诱导增强136 。最后,在细胞周期停滞的癌细胞中,PUFA 可以被引导到脂滴中,促进铁死亡抵抗137 。 因此,在一些细胞中,PUFA 优先隔离在脂滴中,远离膜磷脂,限制了铁死亡的敏感性。
图 4:PUFA 通量和脂滴中的隔离。

Polyunsaturated fatty acids (PUFAs), which may be derived from dietary sources or released from the phospholipids by phospholipases, are conjugated with coenzyme A (CoA) by acyl-CoA synthetase enzymes such as acyl-CoA synthetase long-chain family member 4 (ACSL4), a process known as fatty acid activation, and are then incorporated into triacylglycerol (TAG) that is stored in lipid droplets. Sequestration of PUFAs within TAGs can reduce the amount of PUFAs available to be incorporated into phospholipids and may also protect PUFAs from oxidation. PUFAs esterified within TAG can be released from lipid droplets via lipolysis, which involves a series of lipid droplet-associated lipases, or via lipophagy. The released PUFAs may be incorporated into newly synthesized phospholipids or existing phospholipids (via the Lands cycle), thereby sensitizing membranes to oxidative damage. Lipid droplets may sequester and/or release oxidized PUFAs or their breakdown products to influence ferroptosis. AGPAT, acyl-CoA:acylglycerol phosphate acyltransferase; ATGL, adipose triglyceride lipase; DAG, diacylglycerol; DGAT, acyl-CoA:diacylglycerol acyltransferase; ER, endoplasmic reticulum; G-3-P, glycerol-3-phosphate; GPAT, glycerol phosphate acyltransferase; HSL, hormone-sensitive lipase; LPA, lysophosphatidic acid; MAG, monoacylglycerol; MAGL, monoacylglycerol lipase; PA, phosphatidic acid.
多不饱和脂肪酸 (PUFA) 可能源自膳食来源或通过磷脂酶从磷脂中释放,通过酰基辅酶 A 合成酶(例如酰基辅酶 A 合成酶长链家族成员 4 (ACSL4))与辅酶 A (CoA) 缀合),这一过程称为脂肪酸活化,然后掺入储存在脂滴中的三酰甘油(TAG)中。将 PUFA 封存在 TAG 中可以减少可掺入磷脂中的 PUFA 量,还可以保护 PUFA 免遭氧化。 TAG 内酯化的 PUFA 可以通过脂肪分解(涉及一系列脂滴相关脂肪酶)或通过脂肪自噬从脂滴中释放出来。释放的 PUFA 可能会并入新合成的磷脂或现有的磷脂(通过 Lands 循环),从而使膜对氧化损伤敏感。脂滴可能会隔离和/或释放氧化的PUFA或其分解产物以影响铁死亡。 AGPAT,酰基辅酶A:酰基甘油磷酸酰基转移酶; ATGL,脂肪甘油三酯脂肪酶; DAG,二酰基甘油; DGAT,酰基辅酶A:二酰基甘油酰基转移酶; ER,内质网; G-3-P,3-磷酸甘油; GPAT,磷酸甘油酰基转移酶; HSL,激素敏感脂肪酶; LPA,溶血磷脂酸; MAG,单酰甘油; MAGL,单酰甘油脂肪酶; PA,磷脂酸。
However, lipid droplets do not always seem to protect against ferroptosis. In clear cell renal cell carcinoma cells, hypoxia-inducible factor 2α (HIF2α; also known as EPAS1) regulates the expression of hypoxia-inducible lipid droplet-associated protein (HILPDA). HILPDA inhibits patatin-like phospholipase domain-containing 2 (PNPLA2; also known as ATGL), the rate-limiting enzyme in TAG lipolysis. HILPDA expression increases lipid droplet numbers and PUFA-TAG levels, concomitant with increased ferroptosis sensitivity138. Conversely, HILPDA depletion reduces ferroptosis sensitivity138. Furthermore, system xc– inhibition together with dehydroascorbic acid (that is, oxidized vitamin C) treatment can trigger ferroptosis that involves the oxidation of lipid droplets139. In this scenario, inhibition of diacylglycerol O-acyltransferases (DGATs; the rate-limiting enzymes in TAG synthesis) prevents ferroptosis139. Thus, lipid droplets do not always serve as a protective enclosure that prevents PUFA oxidation. Oxidized TAG species have been measured by lipidomics140, but whether the oxidation of TAGs is common and whether oxidized PUFAs are released from TAGs to propagate ferroptosis is unclear. The mechanisms that influence TAG oxidation are also unknown. Given the localization of FSP1 to lipid droplets one can speculate that FSP1 recycles RTAs at this site to protect TAGs from oxidation, but this possibility remains untested. Furthermore, the mechanisms that determine PUFA sequestration or release via lipolysis or lipophagy are also outstanding questions.
然而,脂滴似乎并不总是能防止铁死亡。在透明细胞肾细胞癌细胞中,缺氧诱导因子 2α(HIF2α;也称为 EPAS1)调节缺氧诱导脂滴相关蛋白(HILPDA)的表达。 HILPDA 抑制马铃薯蛋白样磷脂酶结构域 2 (PNPLA2;也称为 ATGL),这是 TAG脂肪分解中的限速酶。 HILPDA 表达增加脂滴数量和 PUFA-TAG 水平,同时增加铁死亡敏感性138 。相反,HILPDA 耗尽会降低铁死亡敏感性138 。此外,系统 x c –抑制与脱氢抗坏血酸(即氧化维生素 C)治疗一起可以引发涉及脂滴氧化的铁死亡139 。在这种情况下,抑制二酰基甘油 O-酰基转移酶(DGAT;TAG 合成中的限速酶)可防止铁死亡139 。因此,脂滴并不总是充当防止 PUFA 氧化的保护外壳。氧化的 TAG 种类已通过脂质组学测量140 ,但 TAG 的氧化是否常见以及氧化的 PUFA 是否从 TAG 中释放以传播铁死亡尚不清楚。影响 TAG 氧化的机制尚不清楚。鉴于 FSP1 定位于脂滴,我们可以推测 FSP1 在此位点回收 RTA 以保护 TAG 免遭氧化,但这种可能性尚未经过测试。此外,决定 PUFA 通过脂解或脂噬作用封存或释放的机制也是悬而未决的问题。
Finally, in some contexts, lipid droplet formation does not affect ferroptosis sensitivity positively or negatively. For example, in fibrosarcoma cells incubated with extracellular MUFAs, the number of lipid droplets is increased, but disrupting lipid droplet synthesis using small-molecule DGAT1 and DGAT2 inhibitors does not alter ferroptosis sensitivity or the ability of exogenous MUFAs to inhibit ferroptosis28. Inhibition of the DGAT enzymes also has no effect on ferroptosis sensitivity in an osteosarcoma cell line41, further highlighting the context-specific role of lipid droplets in ferroptosis.
最后,在某些情况下,脂滴的形成不会对铁死亡敏感性产生积极或消极的影响。例如,在与细胞外 MUFA 一起孵育的纤维肉瘤细胞中,脂滴数量增加,但使用小分子 DGAT1 和 DGAT2 抑制剂破坏脂滴合成不会改变铁死亡敏感性或外源 MUFA 抑制铁死亡的能力28 。 DGAT 酶的抑制对骨肉瘤细胞系的铁死亡敏感性也没有影响41 ,进一步强调了脂滴在铁死亡中的特定作用。
Peroxisomes 过氧化物酶体
Peroxisomes have important roles in several metabolic processes, including very-long-chain fatty acid breakdown and the catabolism of hydrogen peroxide by catalase. However, peroxisomes are currently best understood to contribute to the regulation of ferroptosis sensitivity via the synthesis of ether phospholipids. PUFA-containing ether phospholipids are abundant in the plasma membrane, and these lipids contribute to ferroptosis execution in some contexts. Ether lipid synthesis is a multistep process that involves collaboration between several compartments of the cell. A key precursor molecule, 1-O-alkyl-glycerol-3-phosphate (AGP), is synthesized in the peroxisomes by FAR1, GNPAT, and AGPS and then shuttled to the ER, where further acylation, head group addition and other modifications can be introduced. Genetic disruption of peroxisome biogenesis or peroxisomal AGP synthesis suppresses ferroptosis in renal cell carcinoma models, hepatocellular carcinoma models and endometrial carcinoma models, concomitant with a reduction in the levels of PUFA-containing ether lipid species25. However, there are also cell-specific differences. For example, deletion of AGPS in fibrosarcoma cells eliminates PUFA-containing ether lipids without altering ferroptosis sensitivity27. Moreover, in Caenorhabditis elegans, disruption of the worm AGPS orthologue (ads-1) actually enhances ferroptosis sensitivity in germ cells101. Here, the protective effect of ADS-1-dependent ether lipid synthesis may be explained by the fact that, in C. elegans (versus mammalian cells), ether lipids are more likely to be acylated with anti-ferroptotic MUFA species than with pro-ferroptotic PUFA species141. These findings highlight how the same metabolic pathway can have different effects on ferroptosis depending on the nature of the final products that are synthesized.
过氧化物酶体在多种代谢过程中具有重要作用,包括超长链脂肪酸分解和过氧化氢酶对过氧化氢的分解代谢。然而,目前最好的理解是,过氧化物酶体通过醚磷脂的合成来调节铁死亡敏感性。质膜中富含含有 PUFA 的醚磷脂,这些脂质在某些情况下有助于铁死亡的执行。醚脂合成是一个多步骤过程,涉及细胞多个区室之间的协作。一个关键的前体分子 1-O-烷基-甘油-3-磷酸 (AGP),由 FAR1、GNPAT 和 AGPS 在过氧化物酶体中合成,然后穿梭到 ER,在那里可以进行进一步的酰化、头基添加和其他修饰。被介绍。过氧化物酶体生物合成或过氧化物酶体 AGP 合成的遗传破坏可抑制肾细胞癌模型、肝细胞癌模型和子宫内膜癌模型中的铁死亡,同时降低含 PUFA 的醚脂质种类25的水平。然而,也存在细胞特异性的差异。例如,纤维肉瘤细胞中AGPS的缺失消除了含有 PUFA 的醚脂质,而不改变铁死亡敏感性27 。此外,在秀丽隐杆线虫中,破坏蠕虫 AGPS 直向同源物 ( ads-1 ) 实际上会增强生殖细胞中铁死亡的敏感性101 。在这里,ADS-1 依赖性醚脂质合成的保护作用可以通过以下事实来解释:在C. 在线虫(相对于哺乳动物细胞)中,醚脂质更可能被抗铁死亡的 MUFA 物种比促铁死亡的 PUFA 物种酰化141 。这些发现强调了相同的代谢途径如何根据合成的最终产品的性质对铁死亡产生不同的影响。
Golgi apparatus 高尔基体
The Golgi apparatus is a complex stack of membranous compartments with a role in protein and vesicle trafficking and lipid metabolism. Disrupting Golgi structure pharmacologically can enhance ferroptosis sensitivity in HeLa and other cancer cells, possibly by increasing intracellular oxidative stress142. Mechanistically, this may involve the disruption of endogenous RTA metabolism. UBIA prenyltransferase domain-containing protein 1 (UBIAD1) undergoes regulated cycling between the ER and Golgi apparatus143. UBIAD1 utilizes geranylgeranyl pyrophosphate in the synthesis of menaquinone 4 from menadione144. Menaquinone 4 (a vitamin K-family metabolite) acts as an endogenous lipophilic RTA that can be recycled by FSP1 and vitamin K epoxide reductase complex subunit 1 (VKORC1; an ER-localized enzyme145) to suppress ferroptosis43,146,147. UBIAD1 is also proposed to directly mediate CoQ synthesis in the Golgi to prevent lipid peroxidation148, which would be important as this could potentially provide a source of non-mitochondrial CoQ in the secretory pathway and plasma membrane that is accessible by FSP1. The relative contributions of UBIAD1-synthesized CoQ148 and STARD7 transport of mitochondrially synthesized CoQ86 to the pool of plasma membrane CoQ remain unknown. Of note, UBIAD1 binds and inhibits the sterol-accelerated degradation of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase in the ER149. Binding of geranylgeranyl pyrophosphate releases UBIAD1 from HMG-CoA reductase and promotes UBIAD1 trafficking to the Golgi149. Thus, UBIAD1-dependent regulation of the mevalonate pathway may indirectly influence de novo synthesis of CoQ in the mitochondria by controlling the synthesis of CoQ precursors (for example, farnesyl pyrophosphate).
高尔基体是一组复杂的膜室,在蛋白质和囊泡运输以及脂质代谢中发挥作用。从药理学上破坏高尔基体结构可以增强 HeLa 和其他癌细胞的铁死亡敏感性,这可能是通过增加细胞内氧化应激142 。从机制上讲,这可能涉及内源性 RTA 代谢的破坏。含 UBIA 异戊烯基转移酶结构域的蛋白 1 (UBIAD1) 在内质网和高尔基体之间经历受调节的循环143 。 UBIAD1 利用香叶基香叶基焦磷酸从甲萘醌144合成甲萘醌 4。 Menaquinone 4(维生素 K 家族代谢物)作为内源性亲脂性 RTA,可被 FSP1 和维生素 K 环氧化物还原酶复合物亚基 1(VKORC1;一种内质网定位酶145 )回收,以抑制铁死亡43 , 146 , 147 。 UBIAD1 还被提议直接介导高尔基体中的 CoQ 合成,以防止脂质过氧化148 ,这很重要,因为这可能会在 FSP1 可访问的分泌途径和质膜中提供非线粒体 CoQ 来源。 UBIAD1 合成的 CoQ 148和 STARD7 将线粒体合成的 CoQ 86转运到质膜 CoQ 库的相对贡献仍然未知。值得注意的是,UBIAD1 结合并抑制 ER 149中 3-羟基-3-甲基戊二酰 (HMG)-CoA 还原酶的甾醇加速降解。 香叶基香叶基焦磷酸的结合从 HMG-CoA 还原酶中释放 UBIAD1,并促进 UBIAD1 运输至高尔基体149 。因此,甲羟戊酸途径的 UBIAD1 依赖性调节可能通过控制 CoQ 前体(例如法呢基焦磷酸)的合成来间接影响线粒体中 CoQ 的从头合成。
Lysosomes 溶酶体
Lipid peroxidation may occur early on in this organelle under ferroptosis-inducing conditions150. Moreover, lysosomal de-acidification generally inhibits ferroptosis150. These effects are likely explained by the role of lysosomes in iron metabolism. Iron can be imported into the cell via the transferrin–transferrin receptor (TFRC) system. Here, Fe3+–transferrin complexes are internalized by TFRC and eventually trafficked to and liberated within the acidic environment of the lysosome. TFRC silencing inhibits ferroptosis, presumably by limiting the uptake of iron into the cell52,53. Iron storage proteins are also degraded in the lysosome. The adaptor protein nuclear receptor coactivator 4 (NCOA4) links iron-loaded ferritin to the lysosomal catabolic machinery through a selective ferritin autophagy pathway known as ferritinophagy; NCOA4 disruption reduces ferroptosis sensitivity by preventing iron retrieval from ferritin and thereby limiting the amount of loosely coordinated (labile) intracellular iron available to participate in redox reactions151,152. Some evidence indicates that NCOA4-mediated ferritin catabolism may be more important for ferroptosis in response to system xc– inhibition than direct GPX4 inhibition153, an example of context-dependent ferroptosis regulation. More broadly, lysosomal catabolic processes are regulated in a multilayered manner by the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2; also known as NFE2L2); one way that loss of NRF2 can enhance ferroptosis sensitivity is by altering lysosome function to increase the pool of labile intracellular iron that can promote lipid peroxidation154.
在铁死亡诱导条件下,脂质过氧化可能在该细胞器的早期发生150 。此外,溶酶体脱酸通常会抑制铁死亡150 。这些效应可能是通过溶酶体在铁代谢中的作用来解释的。铁可以通过转铁蛋白-转铁蛋白受体(TFRC)系统输入细胞。在这里,Fe 3+ -转铁蛋白复合物被 TFRC 内化,最终运输到溶酶体的酸性环境中并在其中释放。 TFRC沉默抑制铁死亡,可能是通过限制细胞对铁的摄取52 、 53 。铁储存蛋白也在溶酶体中被降解。接头蛋白核受体共激活剂 4 (NCOA4) 通过称为铁蛋白自噬的选择性铁蛋白自噬途径将铁负载铁蛋白与溶酶体分解代谢机制连接起来; NCOA4破坏通过阻止铁蛋白从铁蛋白中回收铁来降低铁死亡敏感性,从而限制可参与氧化还原反应的松散协调(不稳定)的细胞内铁的量151 、 152 。一些证据表明,NCOA4 介导的铁蛋白分解代谢对于响应系统 x c抑制的铁死亡可能比直接 GPX4 抑制更重要153 ,这是背景依赖性铁死亡调节的一个例子。 更广泛地说,溶酶体分解代谢过程由转录因子核因子红细胞 2 相关因子 2 (NRF2;也称为 NFE2L2) 以多层方式调节;失去 NRF2 可以增强铁死亡敏感性的一种方法是通过改变溶酶体功能来增加不稳定的细胞内铁库,从而促进脂质过氧化154 。
In addition to regulating iron metabolism, the catabolic function of lysosomes can be engaged either to promote or to prevent ferroptosis. Chaperone-mediated autophagy (CMA) is a degradative process that terminates in lysosomal protein degradation. CMA requires heat shock 70 kDa protein 8 (HSPA8) and the lysosome-associated membrane glycoprotein 2 (LAMP2). System xc– inhibitors can increase LAMP2 expression, the association of HSPA8 and LAMP2 with GPX4, and the degradation of GPX4 via CMA, thereby enhancing ferroptosis155. GPX4 degradation via CMA is reduced by creatine kinase B (CKB), which, acting as a protein kinase, phosphorylates GPX4 at Ser104 to limit GPX4 interaction with HSPA8 and LAMP2 (ref. 156). While CMA of GPX4 tends to promote ferroptosis, the catabolism of other proteins can inhibit ferroptosis. Namely, lysosomal cathepsin D-mediated catabolism of cysteine-rich extracellular proteins such as albumin, followed by cystinosin-mediated export of cystine from the lysosome to the cytosol, can enhance GSH synthesis and inhibit ferroptosis when extracellular free cystine is limiting59. Albumin is abundant outside the cell, potentially providing an important alternative cysteine supply. Within the cell, one response to cysteine deprivation is activation of the integrated stress response, including increased expression of the transcription factor ATF4 (ref. 157). Depletion of lysosomal cystine specifically increases ATF4 expression and reduces ferroptosis sensitivity158.
除了调节铁代谢之外,溶酶体的分解代谢功能还可以促进或预防铁死亡。分子伴侣介导的自噬 (CMA) 是一种以溶酶体蛋白降解为终点的降解过程。 CMA 需要热休克 70 kDa 蛋白 8 (HSPA8) 和溶酶体相关膜糖蛋白 2 (LAMP2)。 System x c –抑制剂可以增加 LAMP2 表达、HSPA8 和 LAMP2 与 GPX4 的结合,以及通过 CMA 降解 GPX4,从而增强铁死亡155 。肌酸激酶 B (CKB) 可减少 GPX4 通过 CMA 的降解,CKB 作为蛋白激酶,在 Ser 104处磷酸化 GPX4,以限制 GPX4 与 HSPA8 和 LAMP2 的相互作用(参考文献156 )。虽然 GPX4 的 CMA 倾向于促进铁死亡,但其他蛋白质的分解代谢可以抑制铁死亡。也就是说,当细胞外游离胱氨酸受到限制时,溶酶体组织蛋白酶 D 介导的富含半胱氨酸的细胞外蛋白(如白蛋白)的分解代谢,然后是胱氨酸介导的胱氨酸从溶酶体输出到细胞质,可以增强 GSH 合成并抑制铁死亡59 。白蛋白在细胞外含量丰富,可能提供重要的替代半胱氨酸供应。在细胞内,对半胱氨酸剥夺的一种反应是整合应激反应的激活,包括转录因子 ATF4 表达的增加(参考文献157 )。溶酶体胱氨酸的消耗会特异性地增加ATF4表达并降低铁死亡敏感性158 。
The surface of the lysosome is an important site where the nutrient-sensing mTORC1 can be activated. In some cells, active mTORC1 promotes ferroptosis by favouring mRNA translation and the consumption of limited intracellular cysteine supplies in the synthesis of proteins rather than GSH90. In other cells, active mTORC1 inhibits ferroptosis by stimulating the translation of GPX4 or the activation of SREBP1, which, as noted above, promotes anti-ferroptotic MUFA synthesis125,159. Which of these pro-ferroptotic and anti-ferroptotic effects dominates at a given time seems likely to vary by cell line and context.
溶酶体表面是营养感应 mTORC1 可以被激活的重要部位。在一些细胞中,活性 mTORC1 通过促进 mRNA 翻译和在蛋白质合成过程中消耗有限的细胞内半胱氨酸(而不是 GSH 90 )来促进铁死亡。在其他细胞中,活性 mTORC1 通过刺激 GPX4 的翻译或 SREBP1 的激活来抑制铁死亡,如上所述,SREBP1 会促进抗铁死亡的 MUFA 合成125 、 159 。在给定时间,促铁死亡和抗铁死亡中哪一个占主导地位似乎可能因细胞系和环境而异。
The nucleus 细胞核
Morphological alterations of the nucleus (for example, karyorrhexis) are defining features of apoptosis160. By contrast, during ferroptosis, the nuclei appear to remain largely intact and DNA damage is neither a defining feature nor a necessary mechanistic event in this process161. The nuclear envelope of mammalian cells is continuous with the ER membrane and includes PUFA-containing phospholipids that may be susceptible to lipid peroxidation162. A functional role for nuclear lipid peroxidation in ferroptosis has not been reported. However, increased levels of the DNA-damage marker 8-oxo-2′-deoxyguanosine (8-oxo-dG) can correlate with the induction of ferroptosis in vivo163. While DNA damage may not contribute to the execution of ferroptosis, one possibility is that peroxidation of nuclear membrane phospholipids leads to the formation of lipid fragments that locally damage DNA during ferroptosis.
细胞核的形态改变(例如核碎裂)是细胞凋亡的定义特征160 。相比之下,在铁死亡过程中,细胞核似乎基本保持完整,DNA 损伤既不是这一过程的决定性特征,也不是必要的机制事件161 。哺乳动物细胞的核膜与内质网膜是连续的,并且包含可能对脂质过氧化反应敏感的含PUFA的磷脂162 。核脂质过氧化在铁死亡中的功能作用尚未报道。然而,DNA 损伤标记 8-oxo-2'-deoxyguanosine (8-oxo-dG) 水平的增加可能与体内铁死亡的诱导相关163 。虽然 DNA 损伤可能不会导致铁死亡的发生,但一种可能性是核膜磷脂的过氧化导致脂质片段的形成,在铁死亡期间局部损伤 DNA。
Within the nucleus, transcription is a major activity and contributes importantly to the regulation of ferroptosis sensitivity. As one might expect from the context-dependent nature of ferroptosis regulation, diverse transcriptional processes occurring in the nucleus help set the ferroptosis threshold of a given cell. A non-exhaustive list of transcription factors and epigenetic regulators that can regulate ferroptosis sensitivity include p53 (refs. 6,147,164), NRF2 (refs. 154,165,166,167,168), NFE2L1 (refs. 130,131), ZEB1 (ref. 169), STAT1 (ref. 170), ATF3 (ref. 171), YAP1 and TAZ50,172, HIF2α138, PPARα173, NUPR1 (ref. 174), MYCN65,175,176, and BAP1 (refs. 7,177). These proteins regulate the expression of genes (for example, SLC7A11, ACSL4, HILPDA, FSP1, NCOA4, LCN2, VKORC1L1 and LRP8) that, in turn, govern ferroptosis sensitivity in different ways, typically by regulating the levels of proteins or metabolites that impinge on membrane lipid peroxidation. Again, context-specific regulation appears to be the rule, with some transcription factors having an important role in ferroptosis regulation in one cell type but not in another178.
在细胞核内,转录是一项主要活动,对铁死亡敏感性的调节发挥着重要作用。正如人们对铁死亡调节的背景依赖性性质所期望的那样,细胞核中发生的多种转录过程有助于设定给定细胞的铁死亡阈值。可以调节铁死亡敏感性的转录因子和表观遗传调节因子的非详尽列表包括 p53(参考文献6、147、164 ) 、NRF2(参考文献154、165、166、167、168 ) 、 NFE2L1 (参考文献130、131 ) )、ZEB1(参考号169 )、STAT1(参考文献170 )、ATF3(参考文献171 )、YAP1 和TAZ 50、172 、HIF2α 138 、PPARα 173 、NUPR1(参考文献174 ) 、 MYCN 65、175、176和 BAP1(参考文献7 ) 177 )。这些蛋白质调节基因(例如SLC7A11 、 ACSL4 、 HILPDA 、 FSP1 、 NCOA4 、 LCN2 、 VKORC1L1和LRP8 )的表达,进而以不同方式控制铁死亡敏感性,通常通过调节影响铁死亡的蛋白质或代谢物的水平。膜脂过氧化作用。同样,上下文特异性调节似乎是规则,一些转录因子在一种细胞类型中的铁死亡调节中具有重要作用,但在另一种细胞类型中则不然178 。
Dynamic and spatially distributed processes shape ferroptosis sensitivity
动态和空间分布的过程塑造铁死亡敏感性
Above, we have largely described the roles of different structures and organelles in ferroptosis regulation in isolation. However, activities in the plasma membrane, cytosol and various intracellular organelles interact with one another (for example, synthesis of CoQ in the mitochondria and Golgi followed by transport to the plasma membrane). Moreover, metabolism is more broadly coordinated by signalling and transcriptional networks in ways that can alter ferroptosis sensitivity. Some additional examples of these interconnections and how they can regulate ferroptosis are highlighted below.
上面,我们已经详细描述了不同结构和细胞器在铁死亡调节中的作用。然而,质膜、细胞质和各种细胞内细胞器中的活动彼此相互作用(例如,线粒体和高尔基体中 CoQ 的合成,然后转运到质膜)。此外,代谢通过信号传导和转录网络进行更广泛的协调,从而改变铁死亡的敏感性。下面重点介绍了这些互连的一些其他示例以及它们如何调节铁死亡。
Lipid metabolism 脂质代谢
Fatty acid synthesis, trafficking and flux help dictate ferroptosis sensitivity by controlling the abundance and localization of more oxidizable versus less oxidizable lipids. These processes involve interactions between plasma membrane transport processes and enzymes and signalling pathways that are distributed throughout the cell. For example, cytosolic acetyl-CoA carboxylase 1 (ACC1) synthesizes malonyl-CoA, which can be used both in the synthesis of long-chain saturated fatty acids by fatty acid synthase (FASN) and in the elongation of imported PUFAs55 into longer-chain-length PUFA species. The desaturation of long-chain saturated fatty acids by SCD1, FADS1 and FADS2 occurs in the ER, which is also the site where fatty acids are activated by ACSL enzymes and incorporated into phospholipids or TAGs by a host of acyltransferases. ACC1, FASN and SCD1 can all regulate ferroptosis sensitivity but seem to do so in different ways in different cells and situations125,179,180,181. Furthermore, signalling networks impact the function of these enzymes. In one example, under conditions of glucose limitation, cytosolic AMPK is activated and can phosphorylate and inhibit ACC1 and ACC2, which ultimately lowers ferroptosis sensitivity by reducing the abundance of oxidizable PUFA-containing membrane phospholipids (for example, PE 18:0/22:4)30,182, perhaps by limiting the malonyl-CoA-dependent elongation of shorter-chain PUFAs. In another example, oncogenic KRAS activation can increase the expression of FASN, which in turn leads to more incorporation of saturated fatty acids and MUFAs into membrane phosphatidylcholines in place of PUFAs, reducing ferroptosis sensitivity107. Likewise, in FLT3-mutant acute myeloid leukaemia, the expression of FASN and SCD1 is increased, resulting in reduced ferroptosis sensitivity176. Outside the context of cancer, how different signalling networks may dynamically regulate lipid metabolism to influence lipid peroxidation and ferroptosis sensitivity is mostly unclear.
脂肪酸合成、运输和通量通过控制易氧化脂质与不易氧化脂质的丰度和定位来帮助决定铁死亡敏感性。这些过程涉及质膜转运过程与分布在整个细胞中的酶和信号传导途径之间的相互作用。例如,胞质乙酰辅酶A羧化酶1(ACC1)合成丙二酰辅酶A,它既可用于脂肪酸合酶(FASN)合成长链饱和脂肪酸,也可用于将输入的PUFA 55延伸成更长的链。链长 PUFA 种类。 SCD1、FADS1 和 FADS2 对长链饱和脂肪酸的去饱和作用发生在 ER 中,这也是脂肪酸被 ACSL 酶激活并被许多酰基转移酶掺入磷脂或 TAG 的位点。 ACC1、FASN 和 SCD1 都可以调节铁死亡敏感性,但在不同的细胞和情况下似乎以不同的方式发挥作用125 , 179 , 180 , 181 。此外,信号网络影响这些酶的功能。在一个例子中,在葡萄糖限制的条件下,胞质 AMPK 被激活,并且可以磷酸化和抑制 ACC1 和 ACC2,这最终通过减少含可氧化 PUFA 的膜磷脂的丰度来降低铁死亡敏感性(例如,PE 18:0/22: 4) 30 , 182 ,可能是通过限制短链 PUFA 的丙二酰辅酶 A 依赖性延伸。 在另一个例子中,致癌的KRAS激活可以增加 FASN 的表达,进而导致更多的饱和脂肪酸和 MUFA 代替 PUFA 掺入膜磷脂酰胆碱中,从而降低铁死亡敏感性107 。同样,在FLT3突变的急性髓性白血病中,FASN 和 SCD1 的表达增加,导致铁死亡敏感性降低176 。在癌症之外,不同的信号网络如何动态调节脂质代谢以影响脂质过氧化和铁死亡敏感性目前尚不清楚。
How lipids are channelled into different classes of membrane phospholipids, to either suppress or enhance ferroptosis, is also not well understood. For example, phosphatidylethanolamine is a key phospholipid that is often oxidized during ferroptosis21. The synthesis of phosphatidylethanolamine involves the transport of phosphatidylserine from the ER to mitochondria at membrane contact sites known as mitochondria-associated membranes183 (Fig. 3b). Within the mitochondria, phosphatidylserine is enzymatically converted into phosphatidylethanolamine and then transported back to the ER, at which point it can traffic to other compartments in the cell (for example, plasma membrane) via the secretory pathway or lipid transfer proteins183 (Fig. 3b). How these trafficking events may contribute to ferroptosis is not clear. In particular, while oxidized phosphatidylethanolamine has been implicated in ferroptosis, its trafficking within the cell has not been directly studied in the context of ferroptosis. Likewise, the flux of PUFAs into TAGs, concomitant with the biogenesis of lipid droplets from the ER, can be protective against ferroptosis in some cases by restricting the incorporation of PUFAs into phospholipids. However, how decisions to channel fatty acids into different lipid species and to traffic phospholipids from one compartment to another are made is not well understood. Additionally, it is possible that cellular decisions about lipid fluxes are made for reasons completely independent of cell death regulation (for example, connected to membrane remodelling events needed for cell division)117,184 and that increased sensitivity or resistance to ferroptosis is a by-product of these changes.
脂质如何被引导到不同类别的膜磷脂中,以抑制或增强铁死亡,也尚不清楚。例如,磷脂酰乙醇胺是一种关键的磷脂,在铁死亡过程中经常被氧化21 。磷脂酰乙醇胺的合成涉及磷脂酰丝氨酸从内质网转运至线粒体的膜接触位点(称为线粒体相关膜183 )(图3b )。在线粒体内,磷脂酰丝氨酸被酶促转化为磷脂酰乙醇胺,然后运回内质网,此时它可以通过分泌途径或脂质转移蛋白183运输到细胞中的其他区室(例如质膜)(图3b) )。这些贩运事件如何导致铁死亡尚不清楚。特别是,虽然氧化磷脂酰乙醇胺与铁死亡有关,但其在细胞内的运输尚未在铁死亡的背景下直接研究。同样,PUFA 流入 TAG 的过程中,伴随着内质网脂滴的生物发生,在某些情况下可以通过限制 PUFA 掺入磷脂来防止铁死亡。然而,如何决定将脂肪酸引导到不同的脂质种类以及将磷脂从一个区室运输到另一个区室的决策尚不清楚。 此外,细胞关于脂质流动的决定可能是出于完全独立于细胞死亡调节的原因(例如,与细胞分裂所需的膜重塑事件有关) 117、184 ,并且对铁死亡的敏感性或抵抗力的增加是一种副反应。这些变化的产物。
Ferroptosis amplification mechanisms
铁死亡扩增机制
Rising plasma membrane lipid peroxidation can trigger feedforward mechanisms that accelerate this process (Fig. 1b). First, lipid peroxidation leads to water entry and cell swelling, increasing plasma membrane tension, which then further activates mechanosensitive Piezo1 and TRP ion channels, accelerating this loop9. Second, lipid peroxidation can stimulate the activity of protein kinase Cβ (PKCβII), which in turn phosphorylates ACSL4 at Thr328, increasing the activity of this lipid metabolic enzyme and enhancing the synthesis of oxidizable PUFA-containing membrane phospholipids185. Where exactly in the cell this pool of ACSL4 resides is not entirely clear; ACSL4 is thought to localize to the ER but a pool of this enzyme may also be found at the plasma membrane and is therefore possibly able to modulate plasma membrane lipid composition directly. Third, plasma membrane lipid peroxidation can cause the protein CAVIN1 to be released from plasma membrane caveolae, allowing it to migrate into the cytosol, where it can bind to and promote the degradation of NRF2 (ref. 186), thereby limiting the protective response mediated by this transcription factor. Fourth, increasing lipid peroxidation can lead to the hyperoxidation of mitochondrial peroxiredoxin 3 (PRDX3), which then translocates to the plasma membrane and inhibits the function of the system xc– cystine–glutamate antiporter, which will tend to increase ferroptosis sensitivity187. These amplification mechanisms may be engaged to overcome the powerful lipid peroxidation defence and membrane repair mechanisms that operate at the plasma membrane, ensuring that the execution of ferroptosis proceeds to completion once this process has been initiated188.
质膜脂质过氧化的增加可以触发前馈机制,加速这一过程(图1b )。首先,脂质过氧化导致水进入和细胞肿胀,增加质膜张力,然后进一步激活机械敏感的 Piezo1 和 TRP 离子通道,加速这一循环9 。其次,脂质过氧化可以刺激蛋白激酶 Cβ (PKCβII) 的活性,进而磷酸化 ACSL4 Thr 328 ,增加这种脂质代谢酶的活性,并增强可氧化的含 PUFA 膜磷脂的合成185 。这个 ACSL4 库到底位于细胞中的哪个位置尚不完全清楚。 ACSL4 被认为定位于内质网,但这种酶的池也可能存在于质膜上,因此可能能够直接调节质膜脂质组成。第三,质膜脂质过氧化可以导致蛋白质CAVIN1从质膜小窝中释放,使其迁移到细胞质中,在那里它可以结合并促进NRF2的降解(参考文献186 ),从而限制介导的保护反应通过该转录因子。第四,脂质过氧化的增加可导致线粒体过氧化还原蛋白 3 (PRDX3) 过度氧化,然后转运至质膜并抑制xc-胱氨酸-谷氨酸逆向转运蛋白系统的功能,这往往会增加铁死亡敏感性187 。 这些放大机制可能用于克服在质膜上运行的强大的脂质过氧化防御和膜修复机制,确保铁死亡的执行在该过程启动后继续完成188 。
Feedback suppression of ferroptosis sensitivity
铁死亡敏感性的反馈抑制
While some mechanisms may serve to accelerate ferroptosis, others operating in parallel will tend to restrain this process. These mechanisms can also be distributed spatially within the cell. For example, in breast cancer cells exposed to GPX4 inhibitors, the pentaspanin protein prominin 2 (PROM2) can be upregulated and stimulate the release of extracellular vesicles containing iron-loaded ferritin from the cell, thereby attenuating lipid peroxidation189. In another example, in tumour-infiltrating neutrophils, signals emanating from cell surface receptors eventually lead to upregulation of aconitate decarboxylase 1 (ACOD1) expression in the mitochondria, which synthesizes the metabolite itaconate. Itaconate can then diffuse out of the mitochondria, bind the NRF2 negative regulator KEAP1 in the cytosol and stabilize NRF2 to suppress ferroptosis190.
虽然某些机制可能会加速铁死亡,但其他并行运作的机制往往会抑制这一过程。这些机制也可以在细胞内空间分布。例如,在暴露于 GPX4 抑制剂的乳腺癌细胞中,五跨膜蛋白 prominin 2 (PROM2) 可以被上调并刺激含有铁负载铁蛋白的细胞外囊泡从细胞中释放,从而减弱脂质过氧化189 。在另一个例子中,在肿瘤浸润性中性粒细胞中,细胞表面受体发出的信号最终导致线粒体中乌头酸脱羧酶 1 (ACOD1) 表达上调,从而合成代谢物衣康酸。然后衣康酸可以扩散出线粒体,结合细胞质中的 NRF2 负调节因子 KEAP1 并稳定 NRF2 以抑制铁死亡190 。
Intracellular trafficking
细胞内运输
Lipid peroxidation occurring within the cell in response to GPX4 inhibition can have a profound effect on the function of the secretory pathway. As noted above, in some cells, GPX4 inhibition stimulates the secretion of extracellular vesicles189. Additionally, in response to the same stimulus, TFRC is depleted from the Golgi apparatus and the endosomal recycling compartment and accumulates at the plasma membrane191. Why TFRC gets ‘stuck’ at the plasma membrane following GPX4 inhibition is unclear, especially since a different cell surface protein, EGFR, traffics normally in response to GPX4 inhibitor treatment191. This TFRC protein redistribution within the cell is a potential ferroptosis molecular marker. Functionally, reduced trafficking of a key iron metabolism-related protein such as TFRC may also tend to reduce ferroptosis sensitivity by limiting iron uptake. The trafficking of other proteins within the secretory pathway, such as the innate immune regulator stimulator of interferon genes (STING)192, is also inhibited following GPX4 inactivation and increased levels of lipid peroxidation. It is unclear whether the inhibition of STING trafficking contributes to ferroptosis execution. However, this example suggests that a pro-ferroptotic stimulus such as GPX4 inactivation can have diverse effects on protein trafficking that are likely to alter cellular responses to other stimuli. Understanding how intracellular protein and lipid transport networks may change over time in response to ferroptotic stress to either reduce or increase ferroptosis sensitivity is an interesting frontier area.
细胞内因 GPX4 抑制而发生的脂质过氧化反应会对分泌途径的功能产生深远的影响。如上所述,在一些细胞中,GPX4 抑制会刺激细胞外囊泡的分泌189 。另外,响应相同的刺激,TFRC从高尔基体和内体再循环室耗尽并在质膜191处积聚。为什么 TFRC 在 GPX4 抑制后“卡在”质膜上尚不清楚,特别是因为不同的细胞表面蛋白 EGFR 响应 GPX4 抑制剂处理而正常运输191 。这种 TFRC 蛋白在细胞内的重新分布是一个潜在的铁死亡分子标记。从功能上讲,减少关键铁代谢相关蛋白(如 TFRC)的运输也可能通过限制铁的摄取来降低铁死亡敏感性。 GPX4 失活和脂质过氧化水平增加后,分泌途径中其他蛋白质的运输,例如干扰素基因的先天免疫调节刺激剂 (STING) 192也会受到抑制。目前尚不清楚 STING 运输的抑制是否有助于铁死亡的执行。然而,这个例子表明,GPX4 失活等促铁死亡刺激可能对蛋白质运输产生多种影响,可能会改变细胞对其他刺激的反应。了解细胞内蛋白质和脂质转运网络如何随时间变化以响应铁死亡应激,从而降低或增加铁死亡敏感性是一个有趣的前沿领域。
Transcription programmes 转录程序
Different transcription factors can induce gene expression programmes that ultimately regulate ferroptosis sensitivity by modulating the function of multiple pathways and organelles in parallel. NRF2 will generally promote ferroptosis resistance by positively regulating the expression of several factors: GSH biosynthetic enzymes that localize to the cytosol130, FSP1 (refs. 165,166), which is required at the plasma membrane, and proteins involved in iron homeostasis found at the lysosome154. However, these mechanisms are not entirely universal. High NRF2 expression can, in some cancer cells, tend to promote ferroptosis sensitivity by upregulating the expression of a plasma membrane-localized transporter (MRP1; encoded by ABCC1) that exports GSH from the cell57. The tumour suppressor protein p53 also seems to be capable of either promoting or inhibiting ferroptosis through various molecular mechanisms, depending on the cell type and even on the nature of the ferroptosis-inducing stimulus6,117,164,193. Likewise, it seems possible that similar competing mechanisms could exist for other transcriptional regulators of ferroptosis. For example, SREBP1 can positively regulate the expression of ER-resident SCD1 (refs. 125,180) but also of cytosolic enzymes involved in malonyl-CoA and NADPH synthesis194. The coordinate expression of these genes may help enable anti-ferroptotic MUFA synthesis but could conceivably also be involved in enhancing ferroptosis in other ways as described above (for example, NADPH-dependent ROS synthesis that initiates ferroptosis). While it is uncomfortable to not have universal models of ferroptosis regulation by individual transcription factors that apply to all cell types and contexts, it does help illustrate the importance and value of detailed investigations of ferroptosis regulation across systems, with careful attention to how ferroptosis is being induced.
不同的转录因子可以诱导基因表达程序,通过并行调节多个途径和细胞器的功能,最终调节铁死亡敏感性。 NRF2 通常会通过正向调节多种因子的表达来促进铁死亡抵抗:定位于细胞质130 的GSH 生物合成酶、质膜所需的 FSP1(参考文献165、166 )以及在 130 发现的参与铁稳态的蛋白质。溶酶体154 .然而,这些机制并不完全通用。在一些癌细胞中,高 NRF2 表达可能会通过上调从细胞输出 GSH 的质膜定位转运蛋白(MRP1;由ABCC1编码)的表达来促进铁死亡敏感性57 。肿瘤抑制蛋白 p53 似乎也能够通过各种分子机制促进或抑制铁死亡,具体取决于细胞类型,甚至取决于铁死亡诱导刺激的性质6 , 117 , 164 , 193 。同样,铁死亡的其他转录调节因子似乎也可能存在类似的竞争机制。例如,SREBP1 可以正向调节 ER 驻留 SCD1 的表达(参考文献125 、 180 ),而且还可以调节参与丙二酰辅酶 A 和 NADPH 合成的胞质酶的表达194 。 这些基因的协调表达可能有助于实现抗铁死亡的 MUFA 合成,但也可能以上述其他方式参与增强铁死亡(例如,启动铁死亡的 NADPH 依赖性 ROS 合成)。虽然没有适用于所有细胞类型和环境的个体转录因子调节铁死亡的通用模型令人不舒服,但它确实有助于说明跨系统详细研究铁死亡调节的重要性和价值,并仔细关注铁死亡是如何发生的。诱发。
Conclusion and perspective
结论与展望
The execution and regulation of ferroptosis are governed by a network of metabolites, enzymes and biochemical pathways that are distributed throughout the cell (Fig. 5). Ferroptosis execution centres on plasma membrane lipid peroxidation and plasma membrane rupture. Major unanswered questions include whether and how lipid peroxidation reactions spread within the cell, how individual lipid species break down in response to peroxidation, and how these events in turn promote (or inhibit) membrane rupture. Intracellular lipid transport is another area where additional investigation could prove fruitful. Beyond STARD7, the role of lipid transport proteins in ferroptosis is largely unexplored. In general, how inter-organelle communication may shape the ferroptosis response remains a frontier area. This communication may take different forms. For example, stable contacts between organelles could provide a means for the direct spread of lipid peroxidation or the sharing of key pro-ferroptotic or anti-ferroptotic metabolites. One specific possibility is that lipid peroxidation could spread between organelles via membrane contact sites, enabling the propagation of ferroptosis within the cell. More broadly, much of the knowledge described here concerning the cell biology of ferroptosis is derived from studies of cultured cancer cells. Normal cells, including more specialized cells such as neurons or microglia195,196, may use distinct mechanisms to execute ferroptosis or to govern sensitivity to this process.
铁死亡的执行和调节是由分布在整个细胞内的代谢物、酶和生化途径网络控制的(图5 )。铁死亡的执行以质膜脂质过氧化和质膜破裂为中心。主要未解答的问题包括脂质过氧化反应是否以及如何在细胞内扩散,单个脂质种类如何响应过氧化而分解,以及这些事件如何反过来促进(或抑制)膜破裂。细胞内脂质转运是额外研究可能取得成果的另一个领域。除了 STARD7 之外,脂质转运蛋白在铁死亡中的作用很大程度上尚未被探索。一般来说,细胞器间通讯如何影响铁死亡反应仍然是一个前沿领域。这种沟通可以采取不同的形式。例如,细胞器之间的稳定接触可以为脂质过氧化的直接扩散或关键的促铁死亡或抗铁死亡代谢物的共享提供手段。一种具体的可能性是,脂质过氧化可以通过膜接触位点在细胞器之间传播,从而使铁死亡在细胞内传播。更广泛地说,这里描述的有关铁死亡细胞生物学的大部分知识都源自对培养癌细胞的研究。正常细胞,包括更特化的细胞,例如神经元或小胶质细胞195、196 ,可以使用不同的机制来执行铁死亡或控制对此过程的敏感性。
图 5:铁死亡调节的细胞图谱。

Cellular sensitivity to ferroptosis is influenced by spatially segregated processes that occur in many organelles. These include processes that regulate fatty acid availability and the incorporation of oxidizable fatty acids into phospholipids, the synthesis and recycling of endogenous antioxidants, the production and metabolism of reactive oxygen species (ROS), and iron metabolism and sequestration. CoQ, coenzyme Q10; MUFA, monounsaturated fatty acid; NFE2L1, nuclear factor erythroid 2-related factor 1; PUFA, polyunsaturated fatty acid; VLCFA, very-long-chain fatty acid; SREBP, sterol regulatory element-binding protein.
细胞对铁死亡的敏感性受到许多细胞器中发生的空间隔离过程的影响。这些包括调节脂肪酸可用性和可氧化脂肪酸掺入磷脂、内源性抗氧化剂的合成和回收、活性氧(ROS)的产生和代谢以及铁代谢和封存的过程。 CoQ,辅酶Q10; MUFA,单不饱和脂肪酸; NFE2L1,核因子红细胞2相关因子1; PUFA,多不饱和脂肪酸; VLCFA,极长链脂肪酸; SREBP,甾醇调节元件结合蛋白。
The sensitivity of the cell to lethal membrane lipid peroxidation is governed by a host of molecules — hundreds of different metabolites and enzymes, along with iron, distributed throughout the plasma membrane, cytosol and the various organelles. It seems possible that every protein, metabolite or biomolecule that can alter iron, lipid or redox metabolism may have a quantitative effect on lipid peroxidation and sensitivity to ferroptosis10. The relative importance of these molecules may be tied to relative expression and abundance, explaining why they vary between different cells and systems10 (Box 3). To understand and manipulate ferroptosis most effectively, it will be important to fully enumerate all relevant species and understand where they act within the cell to modulate ferroptosis. At a higher level of organization, understanding how contact and communication between two or more mammalian cells32,197,198 and how molecules released by other organisms in the environment that act on mammalian cells199,200 can regulate ferroptosis sensitivity will also be important for a full understanding of this mechanism.
细胞对致死性膜脂质过氧化的敏感性受到许多分子的控制——数百种不同的代谢物和酶以及铁,分布在质膜、细胞质和各种细胞器中。似乎每一种可以改变铁、脂质或氧化还原代谢的蛋白质、代谢物或生物分子都可能对脂质过氧化和铁死亡敏感性产生定量影响10 。这些分子的相对重要性可能与相对表达和丰度有关,这解释了为什么它们在不同细胞和系统之间存在差异10 (框3 )。为了最有效地理解和操纵铁死亡,重要的是全面列举所有相关物种并了解它们在细胞内调节铁死亡的作用。在组织的更高层次上,了解两个或多个哺乳动物细胞32 , 197 , 198之间的接触和通讯如何以及环境中其他生物体释放的作用于哺乳动物细胞199 , 200 的分子如何调节铁死亡敏感性也很重要。充分了解这一机制。
Finally, viewing ferroptosis from a cell biological perspective advances our understanding of how we might best promote or interfere with this process by modulating the function of different structures within the cell. Intriguing connections between ferroptosis and other forms of non-apoptotic cell death are emerging at the level of membrane repair, where ESCRT-III complex proteins have roles in negatively regulating multiple forms of cell death. Speculatively, interventions that augment this repair system could delay or prevent multiple forms of cell death at one time, which could be useful for pathologies where more than one form of non-apoptotic cell death is activated. By contrast, agents that can inactivate these pan-pathway protective mechanisms may be especially useful to treat diseases such as cancer, where activation of any one lethal mechanism may be overcome by compensatory mechanisms. Efforts to target ferroptosis therapeutically will be greatly aided by the discovery of additional markers of this process. Recent studies identifying re-localized transferrin receptor and PRDX3 as ferroptosis markers187,191, as well as computer-aided analyses of ferroptotic cell morphology201, highlight the importance of incorporating cell biology into these efforts.
最后,从细胞生物学角度观察铁死亡可以增进我们对如何通过调节细胞内不同结构的功能来最好地促进或干扰这一过程的理解。铁死亡和其他形式的非凋亡细胞死亡之间的有趣联系正在膜修复水平上出现,其中 ESCRT-III 复合蛋白在负调节多种形式的细胞死亡中发挥作用。据推测,增强这一修复系统的干预措施可以同时延迟或防止多种形式的细胞死亡,这对于激活不止一种形式的非凋亡细胞死亡的病理学可能有用。相比之下,能够灭活这些全通路保护机制的药物可能特别适用于治疗癌症等疾病,其中任何一种致死机制的激活都可以通过补偿机制来克服。该过程的其他标记物的发现将极大地帮助以治疗为目标的铁死亡的努力。最近的研究将重新定位的转铁蛋白受体和 PRDX3 鉴定为铁死亡标记物187 、 191 ,以及铁死亡细胞形态的计算机辅助分析201 ,强调了将细胞生物学纳入这些努力的重要性。
References 参考
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
迪克森,SJ 等人。铁死亡:一种铁依赖性非凋亡细胞死亡形式。细胞149 , 1060–1072 (2012)。Galy, B., Conrad, M. & Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis.Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-023-00648-1IF: 81.3 Q1 (2023).
Galy, B.、Conrad, M. 和 Muckenthaler, M. 控制细胞和全身铁稳态的机制。 https://doi.org/10.1038/s41580-023-00648-1如果:81.3 Q1 (2023)。Conrad, M. & Pratt, D. A. The chemical basis of ferroptosis. Nat. Chem. Biol. 15, 1137–1147 (2019).
Conrad, M. & Pratt, DA 铁死亡的化学基础。纳特。化学。生物。 15、1137-1147 (2019)。Ursini, F. & Maiorino, M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic. Biol. Med. 152, 175–185 (2020).
Ursini, F. & Maiorino, M. 脂质过氧化和铁死亡:GSH 和 GPx4 的作用。自由基。生物。医学。 152、175–185 (2020)。Conrad, M. et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 32, 602–619 (2018).
康拉德,M.等人。不同物种脂质过氧化和铁死亡的调节。基因开发。 32、602-619 (2018)。Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
江,L.等人。铁死亡是肿瘤抑制过程中 p53 介导的活性。自然520 , 57–62 (2015)。Zhang, Y. et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 20, 1181–1192 (2018).
张,Y.等人。 BAP1 将铁死亡的代谢调节与肿瘤抑制联系起来。纳特。细胞生物学。 20、1181-1192 (2018)。Stockwell, B. R. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421 (2022).
Stockwell,BR 铁死亡十周年:新兴机制、生理功能和治疗应用。第 185单元,2401–2421 (2022)。Hirata, Y. et al. Lipid peroxidation increases membrane tension, Piezo1 gating, and cation permeability to execute ferroptosis.Curr. Biol. 33, 1282–1294.e5 (2023).
平田,Y.等人。脂质过氧化增加膜张力、Piezo1 门控和阳离子渗透性以执行铁死亡。电流。生物。 33、1282–1294.e5 (2023)。Dixon, S. J. & Pratt, D. A. Ferroptosis: a flexible constellation of related biochemical mechanisms. Mol. Cell 83, 1030–1042 (2023).
Dixon, SJ & Pratt, DA 铁死亡:相关生化机制的灵活组合。摩尔。第83单元,1030–1042 (2023)。Green, D. R. The coming decade of cell death research: five riddles. Cell 177, 1094–1107 (2019).
Green,DR 细胞死亡研究的未来十年:五个谜语。单元177,1094–1107 (2019)。Kayagaki, N. et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136 (2021).
Kayagaki,N.等人。 NINJ1 在裂解细胞死亡过程中介导质膜破裂。自然591 , 131–136 (2021)。Dondelinger, Y. et al. NINJ1 is activated by cell swelling to regulate plasma membrane permeabilization during regulated necrosis. Cell Death Dis. 14, 755 (2023).
唐德林格,Y.等人。 NINJ1 由细胞肿胀激活,以在调节性坏死过程中调节质膜通透性。细胞死亡疾病。 14 , 755 (2023)。Pope, L. E. & Dixon, S. J. Regulation of ferroptosis by lipid metabolism. Trends Cell Biol. 33, 1077–1087 (2023).
Pope, LE 和 Dixon, SJ 通过脂质代谢调节铁死亡。趋势细胞生物学。 33,1077-1087 (2023)。Liang, D., Minikes, A. M. & Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 82, 2215–2227 (2022).
Liang, D.、Minikes, AM 和 Jiang, X. 脂质代谢和细胞信号传导交叉点的铁死亡。摩尔。第82单元,2215–2227 (2022)。Li, Z., Lange, M., Dixon, S. J. & Olzmann, J. A. Lipid quality control and ferroptosis: from concept to mechanism. Annu. Rev. Biochem. https://doi.org/10.1146/annurev-biochem-052521-033527IF: 12.1 Q1 (2023).
Wenzel, S. E. et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628–641.e6 (2017).
温泽尔,SE 等人。 PEBP1 通过使脂氧合酶产生脂质死亡信号来防止铁死亡。单元格171 , 628–641.e6 (2017)。Shah, R., Shchepinov, M. S. & Pratt, D. A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 4, 387–396 (2018).
Shah, R.、Shchepinov, MS 和 Pratt, DA 解决脂氧合酶在铁死亡启动和执行中的作用。 ACS中心。科学。 4、387-396 (2018)。Zilka, O. et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 3, 232–243 (2017).
齐尔卡,O.等人。关于ferrostatin-1和liproxstatin-1的细胞保护机制以及脂质过氧化在铁死亡细胞死亡中的作用。 ACS中心。科学。 3、232-243 (2017)。Anthonymuthu, T. S. et al. Resolving the paradox of ferroptotic cell death: ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 38, 101744 (2021).
安东尼穆图,TS 等人。解决铁死亡的悖论:ferrostatin-1 与 15LOX/PEBP1 复合物结合,抑制过氧化 ETE-PE 的生成,并防止铁死亡。氧化还原生物。 38、101744 (2021)。Tyurina, Y. Y. et al. Redox phospholipidomics discovers pro-ferroptotic death signals in A375 melanoma cells in vitro and in vivo. Redox Biol. 61, 102650 (2023).
Tyurina,YY 等人。氧化还原磷脂组学在体外和体内发现 A375 黑色素瘤细胞中的促铁死亡信号。氧化还原生物。 61、102650 (2023)。Kathman, S. G., Boshart, J., Jing, H. & Cravatt, B. F. Blockade of the lysophosphatidylserine lipase ABHD12 potentiates ferroptosis in cancer cells. ACS Chem. Biol. 15, 871–877 (2020).
Kathman, SG、Boshart, J.、Jing, H. 和 Cravatt, BF 阻断溶血磷脂酰丝氨酸脂肪酶 ABHD12 可增强癌细胞的铁死亡。 ACS 化学。生物。 15 , 871–877 (2020)。Kraft, V. A. N. et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent. Sci. 6, 41–53 (2020).
卡夫、范等人。 GTP 环水解酶 1/四氢生物蝶呤通过脂质重塑对抗铁死亡。 ACS中心。科学。 6、41-53 (2020)。Phadnis, V. V. et al. MMD collaborates with ACSL4 and MBOAT7 to promote polyunsaturated phosphatidylinositol remodeling and susceptibility to ferroptosis. Cell Rep. 42, 113023 (2023).
帕德尼斯,VV 等人。 MMD 与 ACSL4 和 MBOAT7 合作促进多不饱和磷脂酰肌醇重塑和铁死亡的易感性。细胞报告42 , 113023 (2023)。Zou, Y. et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608 (2020).
邹,Y.等人。醚脂质的可塑性促进铁死亡的易感性和逃避。自然585 , 603–608 (2020)。Reed, A., Ware, T., Li, H., Fernando Bazan, J. & Cravatt, B. F. TMEM164 is an acyltransferase that forms ferroptotic C20:4 ether phospholipids. Nat. Chem. Biol. 19, 378–388 (2023).
Reed, A.、Ware, T.、Li, H.、Fernando Bazan, J. 和 Cravatt, BF TMEM164 是一种酰基转移酶,可形成铁焦亡 C20:4 醚磷脂。纳特。化学。生物。 19 , 378–388 (2023)。Magtanong, L. et al. Context-dependent regulation of ferroptosis sensitivity. Cell Chem. Biol. 29, 1409–1418.e6 (2022).
Magtanong,L.等人。铁死亡敏感性的上下文依赖性调节。细胞化学。生物。 29、1409–1418.e6 (2022)。Magtanong, L. et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. 26, 420–432.e9 (2019).
Magtanong,L.等人。外源性单不饱和脂肪酸促进铁死亡抵抗细胞状态。细胞化学。生物。 26 , 420–432.e9 (2019)。von Krusenstiern, A. N. et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat. Chem. Biol. 19, 719–730 (2023).
冯·克鲁森斯蒂恩,AN 等人。铁死亡中脂质过氧化重要位点的鉴定。纳特。化学。生物。 19 , 719–730 (2023)。Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
卡根,VE 等人。氧化的花生四烯酸和肾上腺 PE 引导细胞走向铁死亡。纳特。化学。生物。 13、81-90 (2017)。Pedrera, L. et al. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 28, 1644–1657 (2021).
佩德雷拉,L.等人。铁死亡孔诱导 Ca 2+通量和 ESCRT-III 激活来调节细胞死亡动力学。细胞死亡不同。 28 , 1644–1657 (2021)。Riegman, M. et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 22, 1042–1048 (2020).
里格曼,M.等人。铁死亡通过渗透机制发生,并且独立于细胞破裂而传播。纳特。细胞生物学。 22、1042–1048 (2020)。Agmon, E., Solon, J., Bassereau, P. & Stockwell, B. R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 8, 5155 (2018).
Agmon, E.、Solon, J.、Bassereau, P. 和 Stockwell, BR 模拟铁死亡期间脂质过氧化对膜特性的影响。科学。报告8,5155 (2018)。Roveri, A., Maiorino, M., Nisii, C. & Ursini, F. Purification and characterization of phospholipid hydroperoxide glutathione peroxidase from rat testis mitochondrial membranes. Biochim. Biophys. Acta 1208, 211–221 (1994).
Roveri,A.,Maiorino,M.,Nisii,C. 和 Ursini,F。大鼠睾丸线粒体膜磷脂氢过氧化物谷胱甘肽过氧化物酶的纯化和表征。生物化学。生物物理学。 Acta 1208,211-221 (1994)。Seiler, A. et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248 (2008).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014).
Mao, C. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590 (2021).
Mishima, E. et al. DHODH inhibitors sensitize to ferroptosis by FSP1 inhibition. Nature 619, E9–E18 (2023).
Mao, C., Liu, X., Yan, Y., Olszewski, K. & Gan, B. Reply to: DHODH inhibitors sensitize to ferroptosis by FSP1 inhibition. Nature 619, E19–E23 (2023).
Mao, C., Liu, X., Yan, Y., Olszewski, K. & Gan, B. 回复:DHODH 抑制剂通过抑制 FSP1 对铁死亡敏感。自然619 ,E19–E23(2023)。Labrecque, C. L. & Fuglestad, B. Electrostatic drivers of GPx4 interactions with membrane, lipids, and DNA. Biochemistry 60, 2761–2772 (2021).
Labrecque, CL 和 Fuglestad, B. GPx4 与膜、脂质和 DNA 相互作用的静电驱动因素。生物化学60 , 2761–2772 (2021)。Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Bersuker,K.等人。 CoQ 氧化还原酶 FSP1 与 GPX4 平行发挥作用,抑制铁死亡。自然575 , 688–692 (2019)。Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
多尔,S.等人。 FSP1 是一种不依赖于谷胱甘肽的铁死亡抑制剂。自然575 , 693–698 (2019)。Mishima, E. et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608, 778–783 (2022).
三岛,E.等人。非典型维生素 K 循环是一种有效的铁死亡抑制剂。自然608 , 778–783 (2022)。Lv, Y. et al. Structural insights into FSP1 catalysis and ferroptosis inhibition. Nat. Commun. 14, 5933 (2023).
Lv,Y.等人。 FSP1 催化和铁死亡抑制的结构见解。纳特。交流。 14、5933 (2023)。Soula, M. et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 16, 1351–1360 (2020).
苏拉,M.等人。癌细胞对经典铁死亡诱导剂敏感性的代谢决定因素。纳特。化学。生物。 16、1351-1360 (2020)。Jakaria, M., Belaidi, A. A., Bush, A. I. & Ayton, S. Vitamin A metabolites inhibit ferroptosis. Biomed. Pharmacother. 164, 114930 (2023).
Dai, E., Meng, L., Kang, R., Wang, X. & Tang, D. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem. Biophys. Res. Commun. 522, 415–421 (2020).
Gong, Y. N. et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169, 286–300.e16 (2017).
Ruhl, S. et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 (2018).
Yang, W. H. et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 28, 2501–2508.e4 (2019).
Poursaitidis, I. et al. Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep. 18, 2547–2556 (2017).
Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008).
Gao, M., Monian, P., Quadri, N., Ramasamy, R. & Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308 (2015).
Guan, Y. et al. A single genetic locus controls both expression of DPEP1/CHMP1A and kidney disease development via ferroptosis. Nat. Commun. 12, 5078 (2021).
Ma, X. et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 33, 1001–1012.e5 (2021).
Lin, Z. et al. The lipid flippase SLC47A1 blocks metabolic vulnerability to ferroptosis. Nat. Commun. 13, 7965 (2022).
Cao, J. Y. et al. A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep. 26, 1544–1556.e8 (2019).
Dixon, S. J. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3, e02523 (2014).
Armenta, D. A. et al. Ferroptosis inhibition by lysosome-dependent catabolism of extracellular protein. Cell Chem. Biol. 29, 1588–1600.e7 (2022).
Kobayashi, S. et al. Carnosine dipeptidase II (CNDP2) protects cells under cysteine insufficiency by hydrolyzing glutathione-related peptides. Free Radic. Biol. Med. 174, 12–27 (2021).
Hayashima, K. & Katoh, H. Expression of gamma-glutamyltransferase 1 in glioblastoma cells confers resistance to cystine deprivation-induced ferroptosis. J. Biol. Chem. 298, 101703 (2022).
Zhang, L. et al. Hypersensitivity to ferroptosis in chromophobe RCC is mediated by a glutathione metabolic dependency and cystine import via solute carrier family 7 member 11. Proc. Natl Acad. Sci. USA 119, e2122840119 (2022).
Li, Z. et al. Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat. Chem. Biol. 18, 751–761 (2022).
Greenough, M. A. et al. Selective ferroptosis vulnerability due to familial Alzheimer’s disease presenilin mutations. Cell Death Differ. 29, 2123–2136 (2022).
Alborzinia, H. et al. LRP8-mediated selenocysteine uptake is a targetable vulnerability in MYCN-amplified neuroblastoma. EMBO Mol. Med. 15, e18014 (2023).
Carlisle, A. E. et al. Selenium detoxification is required for cancer-cell survival. Nat. Metab. 2, 603–611 (2020).
Howard, M. T., Carlson, B. A., Anderson, C. B. & Hatfield, D. L. Translational redefinition of UGA codons is regulated by selenium availability. J. Biol. Chem. 288, 19401–19413 (2013).
Harris, I. S. et al. Deubiquitinases maintain protein homeostasis and survival of cancer cells upon glutathione depletion. Cell Metab. 29, 1166–1181.e6 (2019).
Badgley, M. A. et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89 (2020).
Leu, J. I., Murphy, M. E. & George, D. L. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc. Natl Acad. Sci. USA 116, 8390–8396 (2019).
Barayeu, U. et al. Hydropersulfides inhibit lipid peroxidation and ferroptosis by scavenging radicals. Nat. Chem. Biol. 19, 28–37 (2022).
Wu, Z. et al. Hydropersulfides inhibit lipid peroxidation and protect cells from ferroptosis. J. Am. Chem. Soc. 144, 15825–15837 (2022).
Kang, Y. P. et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. 33, 174–189.e7 (2021).
Chen, L. et al. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 1, 404–415 (2019).
Shimada, K., Hayano, M., Pagano, N. C. & Stockwell, B. R. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem. Biol. 23, 225–235 (2016).
Yan, B. et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol. Cell 81, 355–369.e10 (2021).
Zou, Y. et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 16, 302–309 (2020).
Ding, C. C. et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat. Metab. 2, 270–277 (2020).
Nguyen, K. T. et al. The MARCHF6 E3 ubiquitin ligase acts as an NADPH sensor for the regulation of ferroptosis. Nat. Cell Biol. 24, 1239–1251 (2022).
Yagoda, N. et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868 (2007).
Gaschler, M. M. et al. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem. Biol. 13, 1013–1020 (2018).
Bogacz, M. & Krauth-Siegel, R. L. Tryparedoxin peroxidase-deficiency commits trypanosomes to ferroptosis-type cell death. eLife 7, e37503 (2018).
Guo, Y. et al. Small molecule agonist of mitochondrial fusion repairs mitochondrial dysfunction. Nat. Chem. Biol. 19, 468–477 (2023).
Ahola, S. et al. OMA1-mediated integrated stress response protects against ferroptosis in mitochondrial cardiomyopathy. Cell Metab. 34, 1875–1891.e7 (2022).
Guerra, R. M. & Pagliarini, D. J. Coenzyme Q biochemistry and biosynthesis. Trends Biochem. Sci. 48, 463–476 (2023).
Deshwal, S. et al. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat. Cell Biol. 25, 246–257 (2023).
Blomme, A. et al. 2,4-Dienoyl-CoA reductase regulates lipid homeostasis in treatment-resistant prostate cancer. Nat. Commun. 11, 2508 (2020).
Nassar, Z. D. et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. eLife 9, e54166 (2020).
Gao, M. et al. Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363.e3 (2019).
Conlon, M. et al. A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat. Chem. Biol. 17, 665–674 (2021).
Zhang, T. et al. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat. Cancer 3, 75–89 (2022).
Alvarez, S. W. et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643 (2017).
Kalkavan, H. et al. Sublethal cytochrome c release generates drug-tolerant persister cells. Cell 185, 3356–3374.e22 (2022).
Tarangelo, A. et al. Nucleotide biosynthesis links glutathione metabolism to ferroptosis sensitivity. Life Sci. Alliance 5, e202101157 (2022).
Wu, S. et al. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc. Natl Acad. Sci. USA 119, e2121987119 (2022).
Yang, C. et al. De novo pyrimidine biosynthetic complexes support cancer cell proliferation and ferroptosis defence. Nat. Cell Biol. 25, 836–847 (2023).
Wang, F. et al. PALP: a rapid imaging technique for stratifying ferroptosis sensitivity in normal and tumor tissues in situ. Cell Chem. Biol. 29, 157–170.e6 (2022).
Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl Acad. Sci. USA 113, E4966–E4975 (2016).
Ubellacker, J. M. et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118 (2020).
Mann, J. et al. Ferroptosis inhibition by oleic acid mitigates iron-overload-induced injury. Cell Chem. Biol. https://doi.org/10.1016/j.chembiol.2023.10.012IF: 6.6 Q1 (2023).
Perez, M. A., Magtanong, L., Dixon, S. J. & Watts, J. L. Dietary lipids induce ferroptosis in Caenorhabditis elegans and human cancer cells. Dev. Cell 54, 447–454.e4 (2020).
Lee, J. Y. et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc. Natl Acad. Sci. USA 117, 32433–32442 (2020).
Yamane, D. et al. FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem. Biol. 29, 799–810.e4 (2022).
Xin, S. et al. MS4A15 drives ferroptosis resistance through calcium-restricted lipid remodeling. Cell Death Differ. 29, 670–686 (2022).
Dixon, S. J. et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609 (2015).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Bartolacci, C. et al. Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat. Commun. 13, 4327 (2022).
Radif, Y. et al. The endogenous subcellular localisations of the long chain fatty acid-activating enzymes ACSL3 and ACSL4 in sarcoma and breast cancer cells. Mol. Cell Biochem. 448, 275–286 (2018).
Kuch, E. M. et al. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Biophys. Acta 1841, 227–239 (2014).
Poppelreuther, M. et al. The N-terminal region of acyl-CoA synthetase 3 is essential for both the localization on lipid droplets and the function in fatty acid uptake. J. Lipid Res. 53, 888–900 (2012).
Zhu, X. G. et al. CHP1 regulates compartmentalized glycerolipid synthesis by activating GPAT4. Mol. Cell 74, 45–58.e7 (2019).
Beharier, O. et al. PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc. Natl Acad. Sci. USA 117, 27319–27328 (2020).
Sun, W. Y. et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 17, 465–476 (2021).
Chen, D. et al. iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 12, 3644 (2021).
Reed, A. et al. LPCAT3 inhibitors remodel the polyunsaturated phospholipid content of human cells and protect from ferroptosis. ACS Chem. Biol. 17, 1607–1618 (2022).
Liang, D. et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 186, 2748–2794.e22 (2023).
Rodencal, J. et al. Sensitization of cancer cells to ferroptosis coincident with cell cycle arrest. Cell Chem. Biol. https://doi.org/10.1016/j.chembiol.2023.10.011IF: 6.6 Q1 (2023).
Zhao, Y. et al. Identification and characterization of a major liver lysophosphatidylcholine acyltransferase. J. Biol. Chem. 283, 8258–8265 (2008).
Mishra, R. S., Carnevale, K. A. & Cathcart, M. K. iPLA2β: front and center in human monocyte chemotaxis to MCP-1. J. Exp. Med. 205, 347–359 (2008).
Friedmann Angeli, J. P. et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Preprint at Res. Sq. https://doi.org/10.21203/rs.3.rs-943221/v1 (2021).
Yamada, N. et al. DHCR7 as a novel regulator of ferroptosis in hepatocytes. Preprint at bioRxiv https://doi.org/10.1101/2022.06.15.496212 (2022).
Garcia-Bermudez, J. et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122 (2019).
Schumacher, M. M. & DeBose-Boyd, R. A. Posttranslational regulation of HMG CoA reductase, the rate-limiting enzyme in synthesis of cholesterol. Annu. Rev. Biochem. 90, 659–679 (2021).
Conrad, M. & Proneth, B. Selenium: tracing another essential element of ferroptotic cell death. Cell Chem. Biol. 27, 409–419 (2020).
Yi, J., Zhu, J., Wu, J., Thompson, C. B. & Jiang, X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl Acad. Sci. USA 117, 31189–31197 (2020).
Vangala, J. R., Sotzny, F., Kruger, E., Deshaies, R. J. & Radhakrishnan, S. K. Nrf1 can be processed and activated in a proteasome-independent manner. Curr. Biol. 26, R834–R835 (2016).
Tomlin, F. M. et al. Inhibition of NGLY1 inactivates the transcription factor Nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS Cent. Sci. 3, 1143–1155 (2017).
Chavarria, C. et al. ER-trafficking triggers NRF1 ubiquitination to promote its proteolytic activation. iScience 26, 107777 (2023).
Dirac-Svejstrup, A. B. et al. DDI2 Is a ubiquitin-directed endoprotease responsible for cleavage of transcription factor NRF1. Mol. Cell 79, 332–341.e7 (2020).
Forcina, G. C. et al. Ferroptosis regulation by the NGLY1/NFE2L1 pathway. Proc. Natl Acad. Sci. USA 119, e2118646119 (2022).
Kotschi, S. et al. NFE2L1-mediated proteasome function protects from ferroptosis. Mol. Metab. 57, 101436 (2022).
Song, W., Zhang, W., Yue, L. & Lin, W. Revealing the effects of endoplasmic reticulum stress on ferroptosis by two-channel real-time imaging of pH and viscosity. Anal. Chem. 94, 6557–6565 (2022).
Danielli, M., Perne, L., Jarc Jovicic, E. & Petan, T. Lipid droplets and polyunsaturated fatty acid trafficking: balancing life and death. Front. Cell Dev. Biol. 11, 1104725 (2023).
Olzmann, J. A. & Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 20, 137–155 (2019).
Dierge, E. et al. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 33, 1701–1715.e5 (2021).
Minami, J. K. et al. CDKN2A deletion remodels lipid metabolism to prime glioblastoma for ferroptosis. Cancer Cell 41, 1048–1060.e9 (2023).
Lee, H. et al. Cell cycle arrest induces lipid droplet formation and confers ferroptosis resistance. Nat. Commun. 15, 79 (2024).
Zou, Y. et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 10, 1617 (2019).
Ferrada, L., Barahona, M. J., Vera, M., Stockwell, B. R. & Nualart, F. Dehydroascorbic acid sensitizes cancer cells to system xc- inhibition-induced ferroptosis by promoting lipid droplet peroxidation. Cell Death Dis. 14, 637 (2023).
Mohammadyani, D. et al. Molecular speciation and dynamics of oxidized triacylglycerols in lipid droplets: mass spectrometry and coarse-grained simulations. Free Radic. Biol. Med. 76, 53–60 (2014).
Perez, M. A. et al. Ether lipid deficiency disrupts lipid homeostasis leading to ferroptosis sensitivity. PLoS Genet. 18, e1010436 (2022).
Alborzinia, H. et al. Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun. Biol. 1, 210 (2018).
Huang, Y. et al. UBIAD1 alleviates ferroptotic neuronal death by enhancing antioxidative capacity by cooperatively restoring impaired mitochondria and Golgi apparatus upon cerebral ischemic/reperfusion insult. Cell Biosci. 12, 42 (2022).
Nakagawa, K. et al. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature 468, 117–121 (2010).
Rost, S. et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 427, 537–541 (2004).
Jin, D. Y. et al. A genome-wide CRISPR-Cas9 knockout screen identifies FSP1 as the warfarin-resistant vitamin K reductase. Nat. Commun. 14, 828 (2023).
Yang, X. et al. Regulation of VKORC1L1 is critical for p53-mediated tumor suppression through vitamin K metabolism.Cell Metab. 35, 1474–1490.e8 (2023).
Mugoni, V. et al. Ubiad1 is an antioxidant enzyme that regulates eNOS activity by CoQ10 synthesis. Cell 152, 504–518 (2013).
Jo, Y. & DeBose-Boyd, R. A. Post-translational regulation of HMG CoA reductase. Cold Spring Harb. Perspect. Biol. 14, a041253 (2022).
Torii, S. et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 473, 769–777 (2016).
Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016).
Gao, M. et al. Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032 (2016).
Gryzik, M., Asperti, M., Denardo, A., Arosio, P. & Poli, M. NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim. Biophys. Acta Mol. Cell Res. 1868, 118913 (2021).
Anandhan, A. et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci. Adv. 9, eade9585 (2023).
Wu, Z. et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl Acad. Sci. USA 116, 2996–3005 (2019).
Wu, K. et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat. Cell Biol. 25, 714–725 (2023).
Dixon, S. J. & Stockwell, B. R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17 (2014).
Swanda, R. V. et al. Lysosomal cystine governs ferroptosis sensitivity in cancer via cysteine stress response. Mol. Cell 10, 3347–3359.e9 (2023).
Zhang, Y. et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 12, 1589 (2021).
Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 (2007).
Dolma, S., Lessnick, S. L., Hahn, W. C. & Stockwell, B. R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296 (2003).
Vaca, C. E. & Harms-Ringdahl, M. Nuclear membrane lipid peroxidation products bind to nuclear macromolecules. Arch. Biochem. Biophys. 269, 548–554 (1989).
Zhang, Y. et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol. 26, 623–633.e9 (2019).
Tarangelo, A. et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 22, 569–575 (2018).
Muller, F. et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 30, 442–456 (2023).
Koppula, P. et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat. Commun. 13, 2206 (2022).
Takahashi, N. et al. 3D culture models with CRISPR screens reveal hyperactive NRF2 as a prerequisite for spheroid formation via regulation of proliferation and ferroptosis. Mol. Cell 80, 828–844.e6 (2020).
Kuang, F., Liu, J., Xie, Y., Tang, D. & Kang, R. MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem. Biol. 28, 765–775.e5 (2021).
Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017).
Wang, W. et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019).
Wang, L. et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc–. Cell Death Differ. 27, 662–675 (2020).
Wu, J. et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406 (2019).
Venkatesh, D. et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. 34, 526–543 (2020).
Liu, J. et al. NUPR1 is a critical repressor of ferroptosis. Nat. Commun. 12, 647 (2021).
Alborzinia, H. et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat. Cancer 3, 471–485 (2022).
Floros, K. V. et al. MYCN-amplified neuroblastoma is addicted to iron and vulnerable to inhibition of the system Xc–/glutathione axis. Cancer Res. 81, 1896–1908 (2021).
Zhang, Y., Koppula, P. & Gan, B. Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell Cycle 18, 773–783 (2019).
Valente, L. J. et al. p53 deficiency triggers dysregulation of diverse cellular processes in physiological oxygen. J. Cell Biol. 219, e201908212 (2020).
Wohlhieter, C. A. et al. Concurrent mutations in STK11 and KEAP1 promote ferroptosis protection and SCD1 dependence in lung cancer. Cell Rep. 33, 108444 (2020).
Zhao, Y. et al. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep. 33, 108487 (2020).
Tesfay, L. et al. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 79, 5355–5366 (2019).
Lee, H. et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 22, 225–234 (2020).
Acoba, M. G., Senoo, N. & Claypool, S. M. Phospholipid ebb and flow makes mitochondria go. J. Cell Biol. 219, e202023131 (2020).
Barger, S. R., Penfield, L. & Bahmanyar, S. Coupling lipid synthesis with nuclear envelope remodeling. Trends Biochem. Sci. 47, 52–65 (2022).
Zhang, H. L. et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 24, 88–98 (2022).
Wu, Y. et al. Caveolae sense oxidative stress through membrane lipid peroxidation and cytosolic release of CAVIN1 to regulate NRF2. Dev. Cell 58, 376–397.e4 (2023).
Cui, S. et al. Identification of hyperoxidized PRDX3 as a ferroptosis marker reveals ferroptotic damage in chronic liver diseases. Mol. Cell 83, 3931–3939.e5 (2023).
Rodencal, J. & Dixon, S. J. Positive feedback amplifies ferroptosis. Nat. Cell Biol. 24, 4–5 (2022).
Brown, C. W. et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev. Cell 51, 575–586.e4 (2019).
Zhao, Y. et al. Neutrophils resist ferroptosis and promote breast cancer metastasis through aconitate decarboxylase 1. Cell Metab. 35, 1688–1703.e10 (2023).
Feng, H. et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 30, 3411–3423.e7 (2020).
Jia, M. et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 21, 727–735 (2020).
Xie, Y. et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 20, 1692–1704 (2017).
Horton, J. D., Goldstein, J. L. & Brown, M. S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 (2002).
Tian, R. et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci. 24, 1020–1034 (2021).
Ryan, S. K. et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat. Neurosci. 26, 12–26 (2023).
Linkermann, A. et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl Acad. Sci. USA 111, 16836–16841 (2014).
Brown, C. W., Amante, J. J. & Mercurio, A. M. Cell clustering mediated by the adhesion protein PVRL4 is necessary for α6β4 integrin-promoted ferroptosis resistance in matrix-detached cells. J. Biol. Chem. 293, 12741–12748 (2018).
Dar, H. H. et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J. Clin. Invest. 128, 4639–4653 (2018).
Belavgeni, A. et al. vPIF-1 is an insulin-like antiferroptotic viral peptide. Proc. Natl Acad. Sci. USA 120, e2300320120 (2023).
Jin, J. et al. Machine learning classifies ferroptosis and apoptosis cell death modalities with TfR1 immunostaining. ACS Chem. Biol. 17, 654–660 (2022).
Shimada, K. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016).
Murphy, T. H., Miyamoto, M., Sastre, A., Schnaar, R. L. & Coyle, J. T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558 (1989).
Tan, S., Schubert, D. & Maher, P. Oxytosis: a novel form of programmed cell death. Curr. Top. Med. Chem. 1, 497–506 (2001).
Ratan, R. R., Murphy, T. H. & Baraban, J. M. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J. Neurosci. 14, 4385–4392 (1994).
Wiernicki, B. et al. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 11, 922 (2020).
Skouta, R. et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556 (2014).
Poon, J. F., Zilka, O. & Pratt, D. A. Potent ferroptosis inhibitors can catalyze the cross-dismutation of phospholipid-derived peroxyl radicals and hydroperoxyl radicals. J. Am. Chem. Soc. 142, 14331–14342 (2020).
Hadian, K. & Stockwell, B. R. A roadmap to creating ferroptosis-based medicines. Nat. Chem. Biol. 17, 1113–1116 (2021).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Hendricks, J. M. et al. Identification of structurally diverse FSP1 inhibitors that sensitize cancer cells to ferroptosis. Cell Chem. Biol. 30, 1090–1103.e7 (2023).
Nakamura, T. et al. Phase separation of FSP1 promotes ferroptosis. Nature 619, 371–377 (2023).
Farmer, L. A. et al. Intrinsic and extrinsic limitations to the design and optimization of inhibitors of lipid peroxidation and associated cell death. J. Am. Chem. Soc. 144, 14706–14721 (2022).
Jelinek, A. et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 117, 45–57 (2018).
Fang, X. et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl Acad. Sci. USA 116, 2672–2680 (2019).
Merkel, M. et al. Mitochondrial reactive oxygen species formation determines ACSL4/LPCAT2-mediated ferroptosis. Antioxidants 12, 1590 (2023).
Doulias, P. T., Christoforidis, S., Brunk, U. T. & Galaris, D. Endosomal and lysosomal effects of desferrioxamine: protection of HeLa cells from hydrogen peroxide-induced DNA damage and induction of cell-cycle arrest. Free Radic. Biol. Med. 35, 719–728 (2003).
Kurz, T., Gustafsson, B. & Brunk, U. T. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J. 273, 3106–3117 (2006).
Fang, Y. et al. Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: a new mechanism of action. ACS Cent. Sci. 7, 980–989 (2021).
Xu, Y., Li, X., Cheng, Y., Yang, M. & Wang, R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. 34, 16262–16275 (2020).
Li, Y. et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 26, 2284–2299 (2019).
Ide, S. et al. Sex differences in resilience to ferroptosis underlie sexual dimorphism in kidney injury and repair. Cell Rep. 41, 111610 (2022).
Yan, H. et al. Discovery of decreased ferroptosis in male colorectal cancer patients with KRAS mutations. Redox Biol. 62, 102699 (2023).
Jennis, M. et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 30, 918–930 (2016).
Leu, J. I., Murphy, M. E. & George, D. L. Functional interplay among thiol-based redox signaling, metabolism, and ferroptosis unveiled by a genetic variant of TP53. Proc. Natl Acad. Sci. USA 117, 26804–26811 (2020).
Acknowledgements
The authors thank L. Magtanong, D. Pratt and members of the Dixon and Olzmann labs for discussion and comments on the manuscript. This work is supported by the National Institutes of Health (R01GM122923 to S.J.D. and R01GM112948 to J.A.O.) and the American Cancer Society (RSG-21-017-01 to S.J.D. and RSG-19-192-01 to J.A.O.). J.A.O. is a Chan Zuckerberg Biohub Investigator and is also supported by a Bakar Fellows Spark Award.
Ethics declarations
Competing interests
S.J.D. is a co-founder of Prothegen and a member of the scientific advisory board for Hillstream BioPharma. S.J.D. holds patents related to ferroptosis. J.A.O. is a member of the scientific advisory board for Vicinitas Therapeutics and holds patents related to ferroptosis.
Peer review
Peer review information
Nature Reviews Molecular Cell Biology thanks Fudi Wang, José Pedro Friedmann Angeli and Qitao Ran for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Caveolae
-
Small invaginations of the plasma membrane enriched for certain lipids and proteins.
- Endocytosis
-
The process of taking up materials from outside the cells into intracellular vesicles called endosomes. Endosomes can then transport materials within the cell to other compartments such as the lysosome.
- Ether lipid
-
A class of phospholipid where one of the two fatty acyl chains is bound to the glycerol backbone by an ether bond rather than the more common ester bond. Ether lipids may contribute importantly to the execution of ferroptosis in some cells.
- Ferritinophagy
-
The catabolism of iron-laden ferritin nanocages in the autophagolysosome to release free iron atoms.
- Fe–S clusters
-
Iron–sulfur clusters are essential enzyme cofactors. Several different configurations of iron and sulfur atoms yield distinct types of clusters.
- Integrated stress response
-
A gene expression programme that can help the cell respond to a shortage of individual amino acids by increasing the expression of membrane transporters and other metabolic enzymes that restore homeostasis.
- Lipolysis
-
The enzymatic breakdown of triacylglycerol to glycerol and free fatty acids mediated by a series of lipases recruited to the lipid droplet surface.
- Lipophagy
-
The breakdown of the lipid droplet, or a portion of the lipid droplet, through its delivery to the lysosome by a selective autophagic pathway.
- Membrane contact sites
-
Regions of close proximity between two organelles that are often stabilized by protein tethers. These sites typically function as sites for the exchange of lipids and metabolites.
- Monounsaturated fatty acids
-
(MUFAs). Fatty acid molecules that contain a single carbon–carbon double bond.
- Necroptosis and pyroptosis
-
Two forms of non-apoptotic cell death that are biochemically distinct from each other and from ferroptosis.
- Polyunsaturated fatty acid
-
(PUFA). Fatty acid molecule that contains multiple carbon–carbon double bonds, making it more sensitive to oxidative damage.
- Reactive oxygen species
-
(ROS). An umbrella term for a number of small oxygen radicals and species that contain oxygen and which can easily form radical-containing species. Oxygen radicals that may initiate lipid peroxidation, leading to ferroptosis, include the hydroperoxyl radical (the conjugate acid of superoxide) and the hydroxyl radical, which itself can be formed from the Fenton reaction between hydrogen peroxide and iron.
- Selenoprotein
-
One of a small number of proteins in mammalian cells that incorporate the unusual selenium-containing amino acid selenocysteine. Typically, these proteins are involved in some aspect of redox regulation in the cell.
- Stimulator of interferon genes
-
(STING). A protein that can sense DNA in the cytosol and orchestrate a downstream immune response by binding to other signalling proteins.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.

This article is cited by
-
The solute carrier transporters (SLCs) family in nutrient metabolism and ferroptosis
Biomarker Research (2024)
-
Ferroptosis: principles and significance in health and disease
Journal of Hematology & Oncology (2024)
-
Uncovering the role of ferroptosis in Bietti crystalline dystrophy and potential therapeutic strategies
Cell Communication and Signaling (2024)
-
Identification and verification of a novel signature that combines cuproptosis-related genes with ferroptosis-related genes in osteoarthritis using bioinformatics analysis and experimental validation
Arthritis Research & Therapy (2024)
-
Ferroptosis-inducing nanomedicine and targeted short peptide for synergistic treatment of hepatocellular carcinoma
Journal of Nanobiotechnology (2024)